
How titin affects heart growth
It takes about three and a half hours to say the correct chemical name of the giant molecule titin. A Russian journalist demonstrated this fact just over six years ago. So much time is needed because the gigantic, super-long protein is made up of nearly 30,000 amino acids. As a result it is more than one micrometer long, a remarkable length for a single molecule. In the human organism, titin forms the backbone of the sarcomere, which is the smallest functional unit of the muscle.
Titin was as discovered only about 40 years ago. Paradoxically, researchers long overlooked the protein because of its size: The huge protein was simply not mobile enough for gel electrophoresis, a technique used to separate different molecules from each other. It wasn’t until the introduction of highly permeable gels that it became possible to detect titin. After myosin and actin, it is the third most prevalent protein component inside the muscle, where it provides both stability and elasticity because of its special structure.
Many heart diseases can be attributed to defects in titin
Titin’s large size makes its particularly susceptible to mutations.
“Titin’s large size, however, makes its particularly susceptible to mutations,” says Professor Michael Gotthardt, head of the research group on Neuromuscular and Cardiovascular Cell Biology at the Max Delbrück Center for Molecular Medicine (MDC) in Berlin. “We now know that a whole series of human muscle and heart diseases, in particular dilative cardiomyopathy, are caused by a change in the titin molecule.” Dilative cardiomyopathy is a genetic disease in which the cardiac muscle becomes abnormally enlarged, leading to cardiac arrhythmias and increasing incidence of heart failure.
“In the past few years various defects in titin have been discovered using high-throughput sequencing,” explains Gotthardt. “The aim of our newly published study is to determine if a certain change in the molecule allows one to predict which disease will occur – and if perhaps one day it can be treated in a very targeted way.” In addition to Gotthardt and two further MDC researchers who also work at the German Center for Cardiovascular Research in Berlin, colleagues from the University of Arizona in Tucson were also involved in the study.
Deficiencies or changes in titin lead to muscle atrophy

Electron microscopy of a knockout in which the smallest contractive units in the muscle (sarcomeres) fall apart. “On the right you can see the heart of a mouse that only produced a shortened titin, in which the end of the protein can no longer be properly anchored and whose heart was thus reduced in size (atrophic).”
The study’s lead author, Dr. Michael Radke from Gotthardt’s lab, and his colleagues conducted experiments on two groups of mice. In one group, the full knockout (KO) mice, the gene for titin was modified in such a way that the rodents were gradually unable to produce the protein. The other group, the short KO mice, produced a version of the titin molecule in which its M‑band region was truncated in the middle of the sarcomere, but otherwise remained normal.
The researchers reported that all of the gene-modified animals exhibited severe skeletal muscle atrophy at birth. The muscle’s sarcomeres were extremely unstable and could therefore not properly contract. None of the knockout mice lived longer than five weeks. The animals died because their respiratory and cardiac muscles eventually stopped working
The hearts of the mice developed differently
The team observed, however, marked differences in heart development between the full KO and short KO mice. “The complete loss of titin in the full KO mice led to dilative cardiomyopathy, in other words an enlarged heart, which was accompanied by both contraction and filling disturbances,” explains Gotthardt. However, the short KO mice whose titin was truncated developed smaller-than-normal hearts, which to a large extent retained their elastic properties.
“What I found especially astonishing was that fact that the hearts of the full KO mice were, despite the gradual and complete wasting away of the heart muscle’s sarcomeres, apparently still strong enough to grow and to even develop a hypertrophy,” says Gotthardt.
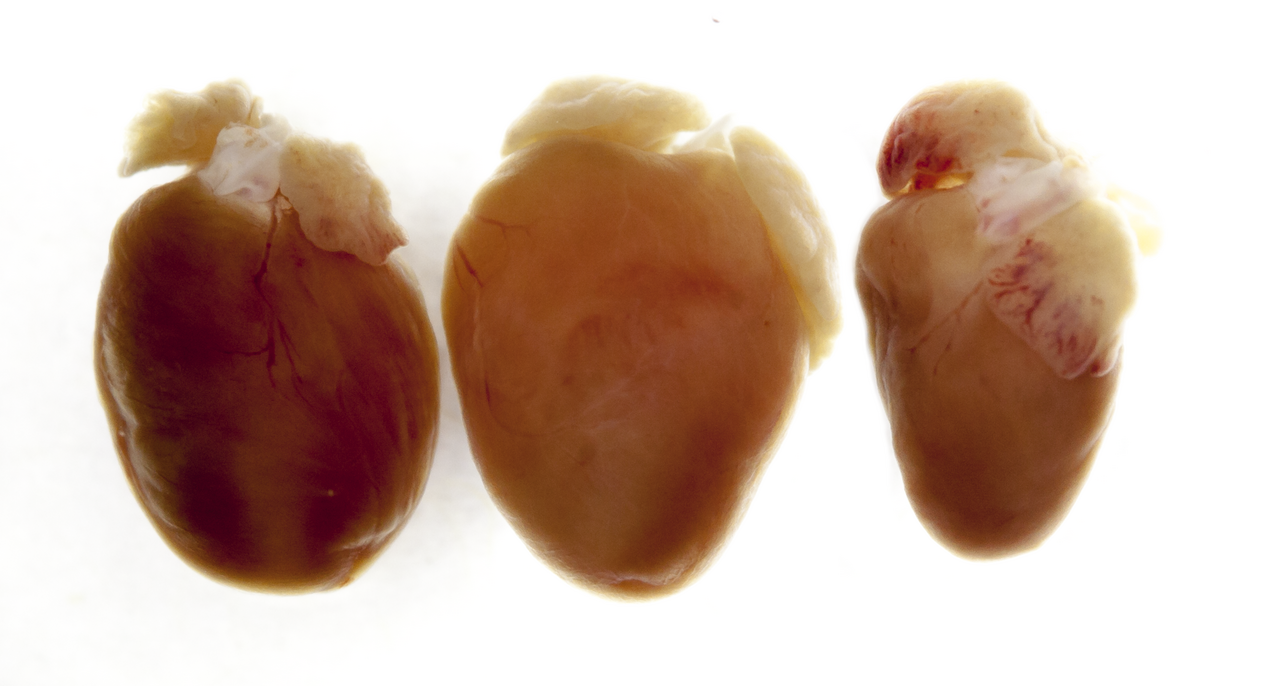
The genetic alteration leaves traces that can even be seen with the naked eye. On the left is a normal heart, in the middle of one of a mouse that could no longer produce titin, thus had an enlarged heart that could no longer fill properly and could not pump properly (dilative cardiomyopathy). On the right you can see the heart of a mouse which only temporarily produced no titin and whose heart was thus reduced in size (atrophic).
Without titin, the body produces more proteins that promote heart growth
In order to better understand how the heart develops differently as the result of titin deficiency or titin truncation and what these differences mean for patients, the research team subsequently determined the number of signaling proteins that are known to be involved in mechanotransduction – i.e., the process in which cells sense and respond to mechanical stimuli by converting them into biochemical signals. This cellular response can lead to changes in the metabolism or growth of the cardiac muscle.
“We discovered that the number of signaling proteins produced in all knockout mice was higher than the number found in healthy animals,” reports Gotthardt, adding, however, that in the case of the full KO mice, who eventually stopped producing titin altogether, the proteins FHL2 and p62 in particular were increasingly synthesized.
FHL2 is a protein that attaches to the anterior elastic part of titin, where the molecule is anchored in the sarcomere like a coiled spring. The p62 protein, on the other hand, operates in a signaling complex on the posterior stiff region of titin, where considerable tension is generated when the cardiac muscle contracts. “These two proteins send out additional growth signals that led to the full KO mice developing enlarged hearts,” says Gotthardt.
The study lays the groundwork for future targeted therapies
The MDC researcher and his team hope the results from their study will help in developing more specific treatments for heart diseases caused by titin defects. “On the one hand, the research could translate into physicians being able to better diagnose and distinguish between the various titinopathies,” says Gotthardt. “On the other hand, it will hopefully one day be possible to tailor treatments of these diseases to the individual patient.”
A gene therapy that would involve removing – and possibly replacing – the defective section of the giant molecule’s blueprint is just one option. The other option, according to Gotthardt, would be to use small molecules to precisely alter the signaling pathways that lead to abnormal heart enlargement.
Further Information

Literature
Michael H. Radke et al. (2019): „Deleting Full Length Titin Versus the Titin M‑Band Region Leads to Differential Mechanosignaling and Cardiac Phenotypes“. Circulation. doi:10.1161/CIRCULATIONAHA.118.037588



