
© AG Hammes-Lewin
Hammes Lab
Molecular Pathways in Cortical Development
Profile
We study developmental processes to understand human health and disease.
Research in the lab is broadly focused on understanding the mechanisms of growth factor and morphogen signaling in development and disease. Our objective is to investigate the pathomechanisms that lead to disorders affecting the developing brain, craniofacial tissue, and heart. Our team aims at understanding how regulatory mechanisms in morphogen signaling, along with mechanical cues, affect the fate decisions of progenitor cells, cell movements, and complex tissue rearrangements. Errors in such tissue and cell choreography result in developmental disorders, so called birth defects. Morphogens and their downstream signaling pathways play critical roles in embryonic development, and pathogenic mutations, such as those in the sonic hedgehog (SHH) pathway, are common genetic causes of human developmental disorders affecting the brain. To understand the disease’s origin, we use a multi-layered experimental approach that combines mouse genetics, high and super resolution imaging, transcriptomic/proteomic approaches and functional assays in human iPSC derived neuronal cell cultures. Analyzing developmental processes not only aids in understanding congenital disorders, but it also sheds light on developmental mechanisms that are “reused” in regeneration and “misused” in cancer. The long-term goal of our research is to use insights gained from developmental mechanisms in the treatment of developmental disorders as well as of childhood and adult cancers.

Team








Research Projects
Our objective is to investigate the pathomechanisms that lead to disorders affecting the developing brain, craniofacial tissue, and heart. In specific, we aim at understanding how regulatory mechanisms in growth factor and morphogen signaling, along with mechanical cues, affect the fate decisions of progenitor cells, cell movements, and complex tissue rearrangements.
- RESEARCH BACKGROUND
- Developmental disorders, so called birth defects, are collectively the leading cause of infant mortality. Even when birth defects can be treated surgically, patients often suffer from associated life-long health problems. Among the most common and debilitating defects are those affecting the formation of the neural tube and the heart. Neural tube defects (NTDs) arise from a complex combination of genetic and environmental interactions. Substantial advances have been made in the prevention and treatment of these disorders. However, NTDs remain a serious public health problem and understanding the etiology of such complex diseases still presents a major challenge. Birth defects often show a variable penetrance of the defects or a variable spectrum of phenotype severity even amongst family members with the same mutations. Modifier genes are major contributors to this variability, but are poorly understood. Another challenge, even in well characterized monogenetic syndromes, is the appearance of comorbidities with unknown etiology in addition to the main birth defect. Yet unknown functions of the protein associated with the disease can be responsible for such defects and insights into the multifunctional aspects of disease associated genes/factors is of major clinical importance.
-

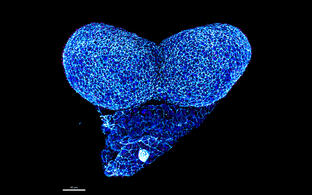
Confocal image of a whole mount neurulation stage mouse embryo. View onto the developing forebrain
- RESEARCH SUMMARY
- Our team studies regulatory mechanisms in morphogen signaling and their interplay with mechanical cues that guide progenitor cell fate decisions, cell movements and complex tissue rearrangements. Failure to execute these orchestrated cell behaviors in space and time results in developmental disorders. We seek to understand the pathomechanisms that lead to birth defects. Uptake of morphogens and their downstream signaling pathways play pivotal roles in embryonic development. Pathogenic mutations e.g., in the sonic hedgehog (SHH) pathway are amongst the most common genetic causes of human developmental disorders affecting the brain. To elucidate disease etiology, we take a multi-layered experimental approach combining mouse genetics, super resolution imaging and functional assays in cell culture, including human induced pluripotent stem cells (hiPSCs). We recently identified disease relevant modifier genes that modulate SHH signaling capacity thus influencing the penetrance and manifestation of forebrain defects. In a recent study we also elucidated mechanisms involved in balancing WNT signaling during early forebrain development in health and disease. Our results highlight the complexity and heterogeneity of mechanisms important to balance SHH and WNT signaling capacity. Properly titrated morphogen activity is crucial for normal patterning and morphogenesis during embryonic development but also for tissue homoeostasis in the adult. We further demonstrated that receptor-mediated endocytosis also orchestrates remodeling events in neuroepithelial cells. Thus we showed that receptor-mediated endocytosis is necessary for the reorganization of apical plasma membranes, cell-cell junctions and cytoskeleton during neural tube closure and neural crest migration. Recent results from our lab indicate that not only morphogen cues needed to govern cell behaviors, but also that cell mechanosensitivity is important in several cell types in the developing brain and heart. Mouse models carrying human pathogenic variants of a mechanosensitive ion channel, display novel developmental cardiovascular and brain phenotypes.
-

Scanning electron microscopy image of the developing forebrain neuroepithelium

- Intricate balance of SHH pathway modulation — a decisive switch between health and disease
Background and previous work in the lab
Identification of disease-relevant modulators of the SHH pathway in the developing brain
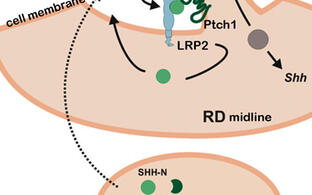
Pathogenic gene variants in humans, affecting the sonic hedgehog (SHH) pathway, lead to severe brain and craniofacial malformations often co-existing with cardiovascular defects with variable penetrance due to unknown genetic modifiers. To identify such modifiers, we established LDL receptor related protein 2 (Lrp2) congenic null mutant mouse models on two different inbred backgrounds. LRP2 is a SHH co-receptor and C57BL/6N mice deficient for this receptor suffer from heart outflow tract defects and holoprosencephaly (HPE), caused by impaired SHH activity in the ventral forebrain as shown by our lab. In Donni-Barrow patients, autosomal recessive LRP2 gene defects are also associated with heart anomalies, craniofacial anomalies, and forebrain defects including holoprosencephaly with inter- and intrafamilial phenotypic variability, a general feature of HPE. Strikingly, SHH signaling and HPE as well as heart defects are 100% penetrant in our Lrp2-/-C57BL/6N mice, but are fully rescued on a congenic FVB/N strain background indicating a strong influence of modifier genes. Applying comparative transcriptomics we identified pituitary tumor transforming gene 1 (Pttg1) and unc-51-like kinase 4 (Ulk4) as candidate genetic modifiers upregulated in the rescue strain. Functional analyses showed that ULK4 and PTTG1 are new positive regulators of SHH signaling at the primary cilium, rendering the pathway more resilient to disturbances (Mecklenburg et al., Development, 2021). The identification of genes, that powerfully modulate the penetrance of genetic disturbances affecting the brain and heart, is directly relevant to understand variability in human developmental disorders.
Ongoing research projects and future perspectives
Imbalance in the morphogen activity, in particular SHH signaling, either predisposes or causes disease such as birth defects or cancer. The mechanisms underlying the complexity and heterogeneity of pathway regulation between individuals are poorly understood. We are currently analyzing the mechanisms underlying the promoting effect on SHH signaling of our previously identified candidate modifiers (Mecklenburg et al., Development, 2021) using primary neuroepithelial cells from different mouse strains and human neuronal cultures derived from induced pluripotent stem cells (hiPSCs). To analyze the complexity and heterogeneity of SHH pathway regulation in health and disease conditions will provide further insight into disease etiology of the primary congenital defects and of possible so far underestimated comorbidities in child- or adulthood.

Mouse and human cell culture models to analyze the complexity of SHH pathway regulation.
A: Primary mouse neuroepithelial stem cells. B: Human neuronal precursor cells (NPCs) C: Differentiated neurons derived from hiPSCs
- (Patho)mechanisms of vertebrate head development: the role of receptor-mediated endocytosis in linking brain and craniofacial morphogenesis by directing neural versus neural crest fate
Background and previous work in the lab
Neural tube closure requires the endocytic receptor LRP2 and its functional interaction with intracellular scaffolds
Pathogenic mutations in the endocytic receptor LRP2 in humans are not only associated with holoprosencephaly, but also with severe neural tube closure defects (NTDs) such as anencephaly and spina bifida. In collaboration with the laboratories of Kerstin Feistel (University Hohenheim, Germany) and John Wallingford (University of Texas at Austin, USA) we have combined neural tube closure analyses in mouse and in Xenopus laevis to elucidate the etiology of LRP2-related NTDs. Loss of LRP2 impaired dorsal neuroepithelial morphogenesis, culminating in NTDs that impeded anterior neural plate folding and neural tube closure in both model organisms. Loss of LRP2 severely impaired plasma membrane remodeling and apical constriction as well as proper localization of the core planar cell polarity (PCP) proteins, ultimately leading to neural tube closure defects (Kowalczyk et al., Development, 2021).
Ongoing research projects and future perspectives
Receptor-mediated endocytosis in directing neural stem cell versus neural crest cell fate
During neurulation, the dorsolateral neural plate border (NPB) not only plays an important role in neural plate elevation and neural tube closure processes, but is also the niche for the cranial neural crest cells (NCCs). NCCs are collectively migrating cells, derived from the neuroepithelium through an epithelial-to-mesenchymal transition (EMT) process and capable of transforming into multiple cell lineages at designated locations. Cranial NCCs produce the craniofacial mesenchyme that differentiates into the cartilage, bone, cranial neurons, glia, and connective tissues of the face. Dysregulated NCC integrities can lead to congenital anomalies so called neurocristopathies in newborns. We have previously detected the endocytic receptor LRP2 as a critical factor for NCC dynamics during early embryonic development. Neural crest cells and many other embryonic and metastatic cell types undergo EMT to migrate long distances to reach their target. Identifying (patho)mechanisms for EMT and directed collective cell migration not only helps to understand processes during embryonic development, but also contributes to understanding EMT and cell migration in disease as well as tissue remodeling in the adult organism.

Scanning electron microscopy image of a neurulation stage mouse embryo and confocal images of delaminating and migrating neural crest cells (NCCs)
- Directed cell migration/movement in tissue formation: Do mechanical guidance cues cooperate with chemotaxis during embryonic development and regeneration in the brain and the cardiovascular system?
Mechanical forces such as stretch, or a stiffness gradient of the extracellular matrix can be sensed by ion channels and thereby trigger signaling pathways and cytoskeletal remodeling important for cell polarity, motility and migration, processes that are crucial for patterning and morphogenesis.
We hypothesize that the mechanosensitive ion channels are relevant during embryonic morphogenesis and in the adult organism during tissue remodeling and regeneration e.g., after ischemic tissue damage. Mechanisms underlying developmental morphogenetic processes are often repeated during regeneration in the adult organism. Identifying the mechanisms underlying mechanosensitive guidance cues during development will lead to a better understanding not only of congenital arterial pathologies but also of the cellular dynamics during regeneration in arterial disease with adult onset.

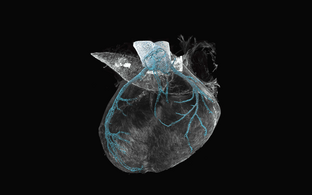
Genetic fate mapping for a mechanosensitive ion channel in the developing mouse heart coronary arteries



Publications
News





More news
We welcome our new students!
Nella Delva is a Fulbright student from the USA who joined our lab in September 2024!
Linda Haazen started on the 1st of October 2024 and is a Master student from the Netherlands.
Congratulations Nihan! Nihan won the poster prize at the ISDN (International Society for Developmental Neuroscience) meeting in Montpellier in September 2024!
Nella Delva is a Fulbright student from the USA who joined our lab in September 2024!
Linda Haazen started on the 1st of October 2024 and is a Master student from the Netherlands.
Congratulations Nihan! Nihan won the poster prize at the ISDN (International Society for Developmental Neuroscience) meeting in Montpellier in September 2024!
