
Noncompaction
Sarcomere Genes in Left Ventricular Noncompaction Cardiomyopathy

Left ventricular noncompaction cardiomyopathy (LVNC) is seen in a number of genetic syndromes and has been associated with neuromuscular disorders and with mitochondrial disease. It is classified as a distinct primary cardiomyopathy with a genetic etiology and may be an isolated finding or may also be associated with other forms of structural congenital heart disease (Jenni et al., 2001). Among many sporadic cases, familial recurrence with autosomal dominant inheritance is observed in isolated LVNC (Sasse-Klaassen et al., 2003).
The large number of mouse models that exhibit LVNC have been thought to represent a generic form of myocardial growth failure resulting from primary cardiac disease as well as from systemic growth disruption (Chen et al., 2009). There are discrete differentiation and maintenance programs for cardiomyocyte subpopulations within compact and trabecular myocardium (de la Pompa et al., 2012). LVNC has been proposed to result from either a reduction in the compact layer or a failure of maturation of the trabecular layer, but to date the primary mechanism remains ill-defined.
In humans, LVNC is morphologically characterized by a severely thickened two-layered myocardium consisting of a thin compacted epicardial layer and a thick noncompacted endocardial layer. The endocardial layer contains numerous prominent trabeculations and deep intertrabecular recesses (Fig.1, Fig.2).
We hypothesized that mutations in sarcomere protein genes may be associated with LVNC. In the first study (Klaassen et al., 2008) mutational analysis in a large cohort of 63 unrelated adult probands with LVNC and absence of other congenital heart anomalies was performed. Genes encoding 6 sarcomere proteins were studied. 11 mutations were identified in 3 distinct genes: β‑myosin heavy chain (MYH7), α‑cardiac actin (ACTC), and Troponin T (TNNT2). Nine distinct mutations, 7 of them in MYH7, 1 in ACTC, and 1 in TNNT2were found. LVNC is within the diverse spectrum of cardiac morphologies triggered by sarcomere protein gene defects.
In our second study we found that heterozygous mutations in genes encoding β‑myosin heavy chain (MYH7), α‑cardiac actin (ACTC1), cardiac troponin T (TNNT2),cardiac myosin-binding protein C (MYBPC3), and alpha-tropomyosin (TPM1) account for 30% of cases of isolated LVNC in adult patients (Probst et al., 2011). The distribution of disease genes confirms genetic heterogeneity and opens new perspectives in genetic testing in patients with LVNC and their relatives at high risk of inheriting the cardiomyopathy. The presence or absence of a sarcomere gene mutation in LVNC could not be related to the clinical phenotype.
In patients with LVNC, heart failure New York Heart Association class III or greater and cardiovascular complications at presentation were strong predictors for adverse outcome (Greutmann et al., 2012). In another study the aim was to find improved quantitative CMR criteria to distinguish LVNC from dilated cardiomyopathy (DCM) and hypertrophic cardiomyopathy (HCM) with high sensitivity and specificity. Absolute cardiac magnetic resonance (CMR) quantification of the total LV myocardial mass index (LVMMI) non-compacted or the percentage LV-MMnon-compacted and increased trabeculation in basal segments allows to reliably diagnose LVNC and to differentiate it from other cardiomyopathies (Grothoff et al., 2012).
Publikationen
Mutations in sarcomere protein genes in left ventricular noncompactionMutations in sarcomere protein genes in left ventricular noncompaction
Sarcomere gene mutations in isolated left ventricular noncompaction cardiomyopathy do not predict clinical phenotypeSarcomere gene mutations in isolated left ventricular noncompaction cardiomyopathy do not predict clinical phenotype
Predictors of adverse outcome in adolescents and adults with isolated left ventricular noncompactionPredictors of adverse outcome in adolescents and adults with isolated left ventricular noncompaction
Value of cardiovascular MR in diagnosing left ventricular non-compaction cardiomyopathy and in discriminating between other cardiomyopathiesValue of cardiovascular MR in diagnosing left ventricular non-compaction cardiomyopathy and in discriminating between other cardiomyopathies
Pressemitteilungen
Mutationen lösen schwere Herzmuskelerkrankung aushttps://www.mdc-berlin.de/12620043/de/news/archive/2008/20080604-mutati…
http://www.escardio.org/communities/Working-Groups/cmp/Documents/WGMP-Newsletter-Oct08-part‑2.pdf
http://circ.ahajournals.org/content/117/22/2847.full.pdf
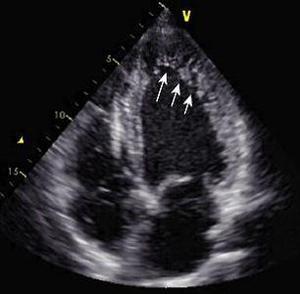
Fig. 2 Echocardiogram of a Patient with LVNC; arrows depict the noncompacted myocardium
Fig. 2 Echocardiogram of a Patient with LVNC; arrows depict the noncompacted myocardium