
Wie Titin das Herzwachstum beeinflusst
Rund dreieinhalb Stunden dauert es, um die korrekte chemische Bezeichnung des Riesenmoleküls Titin auszusprechen. Ein russischer Journalist hat das vor gut sechs Jahren einmal demonstriert. Denn das gigantische, lang gestreckte Protein setzt sich aus fast 30.000 Aminosäuren zusammen. So kommt es auf die für ein einzelnes Molekül beachtliche Länge von mehr als einem Mikrometer. Im menschlichen Organismus stellt Titin das Rückgrat des Sarkomers dar, der kleinsten funktionellen Einheit des Muskels.
Bekannt ist Titin erst seit rund vierzig Jahren. Paradoxerweise wurde das Protein gerade aufgrund seiner Größe lange Zeit übersehen: Für eine Gel-Elektrophorese, die man benutzt, um verschiedene Moleküle voneinander zu trennen, war der Riese schlicht nicht mobil genug. Erst mit der Einführung extrem durchlässiger Gele ließ sich Titin schließlich nachweisen. Im Muskel ist es nach Myosin und Aktin das dritthäufigste Eiweiß und sorgt dort aufgrund seiner besonderen Struktur sowohl für Stabilität als auch für Elastizität.
Viele Herzkrankheiten gehen auf Defekte im Titin zurück
Wegen seiner Größe ist Titin besonders anfällig für Mutationen.
„Wegen seiner Größe ist Titin allerdings auch besonders anfällig für Mutationen“, sagt Professor Michael Gotthardt, der Leiter der Arbeitsgruppe „Neuromuskuläre und kardiovaskuläre Zellbiologie“ am Max-Delbrück-Centrum für Molekulare Medizin (MDC) in Berlin. „Wir wissen inzwischen, dass eine ganze Reihe menschlicher Muskel- und Herzerkrankungen, insbesondere die dilatative Kardiomyopathie, auf eine Veränderung im Titin-Molekül zurückzuführen ist.“ Bei diesem erblichen Leiden ist der Herzmuskel krankhaft erweitert, was zu Herzrhythmusstörungen und zunehmender Herzschwäche führt.
„In den vergangenen Jahren konnten mittels Hochdurchsatz-Sequenzierung unterschiedliche Defekte im Titin aufgespürt werden“, berichtet Gotthardt. „Mit unserer jetzt veröffentlichten Studie wollten wir untersuchen, ob sich anhand einer bestimmten Veränderung in dem Molekül vorhersagen lässt, welche Krankheit aus ihr resultiert – und ob sie sich eines Tages vielleicht ganz spezifisch behandeln lässt.“ Neben Gotthardt und zwei weiteren MDC-Forschern, die auch am Deutschen Zentrum für Herz-Kreislauf-Forschung in Berlin arbeiten, waren an der aktuellen Arbeit Kolleginnen und Kollegen der University of Arizona in Tucson beteiligt.
Fehlendes oder verändertes Titin führte zu Muskelschwäche

Electron microscopy of a knockout in which the smallest contractive units in the muscle (sarcomeres) fall apart. "On the right you can see the heart of a mouse that only produced a shortened titin, in which the end of the protein can no longer be properly anchored and whose heart was thus reduced in size (atrophic)."
Der Erstautor der Publikation, Dr. Michael Radke aus Gotthardts Arbeitsgruppe, und seine Kolleginnen und Kollegen experimentierten mit zwei unterschiedlichen Gruppen von Mäusen. Bei der einen Sorte von Tieren, den Voll-Knock-out-Mäusen, war das Gen für Titin so verändert, dass die Nager das Eiweiß nach und nach nicht mehr herstellen konnten. Die andere Sorte, die Kurz-Knock-out-Mäuse, stellte Titin-Moleküle her, die im Bereich der M-Scheibe in der Mitte des Sarkomers verkürzt waren, ansonsten aber normal aussahen.
Wie die Forscherinnen und Forscher berichten, wiesen alle der genveränderten Tiere von Geburt an eine schwere Skelettmuskelschwäche auf. Die Sarkomere ihrer Muskeln waren extrem instabil und konnten dadurch nicht richtig kontrahieren. Keine der Knock-out-(KO-)Mäuse lebte länger als fünf Wochen. Die Tiere starben, weil ihre Atem- und ihre Herzmuskulatur schließlich versagten.
Die Herzen der Mäuse entwickelten sich unterschiedlich
Deutliche Unterschiede zwischen den Voll-KO- und den Kurz-KO-Mäusen beobachtete das Team um Radke und Gotthardt jedoch bei der Entwicklung des Herzens. „Der komplette Verlust des Titins bei den Voll-KO-Mäusen führte zu dilatativer Kardiomyopathie, also zu einer Vergrößerung des Herzens, die sowohl mit Kontraktions- als auch mit Füllungsstörungen einherging“, erläutert Gotthardt. Bei den Kurz-KO-Mäusen mit verkürztem Titin hingegen waren die Herzen verkleinert, behielten aber insbesondere ihre elastischen Eigenschaften weitgehend bei.
„Besonders erstaunlich fand ich die Tatsache, dass die Herzen der Voll-KO-Mäuse, obwohl die Sarkomere des Herzmuskels nach und nach komplett abgebaut wurden, offenbar noch kräftig genug waren, um zu wachsen und sogar eine Hypertrophie zu entwickeln, also zu groß zu werden“, sagt Gotthardt.
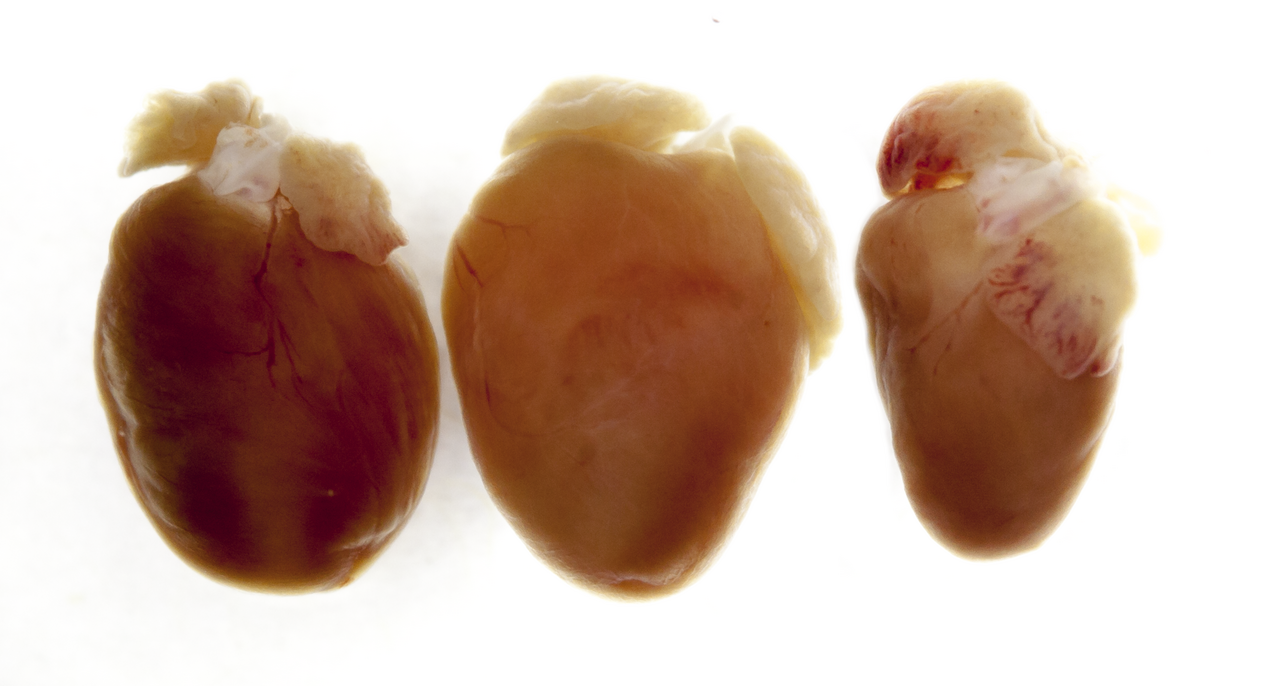
Die Genveränderung hinterlässt Spuren, die selbst mit dem bloßen Auge erkennbar sind. Links ist ein normales Herz zu sehen, in der Mitte eines von Mäusen, die kein Titin mehr herstellen konnten und so ein vergrößertes Herz hatten, das sich nicht mehr richtig füllen und nicht richtig pumpen konnte (dilative Kardiomyopathie). Rechts sieht man das Herz von Mäusen, die nur ein verkürzetes Titin herstellten, bei dem das Ende nicht mehr richtig verankert werden kann und deren Herz dadurch verkleinert war (atroph)
Ohne Titin werden mehr Proteine gebildet, die das Herz wachsen lassen
Die unterschiedliche Entwicklung der Herzen durch fehlendes oder verkürztes Titin und die Bedeutung dieser Unterschiede für Patienten wollte die Forschungsgruppe besser verstehen. Deshalb bestimmte sie im Anschluss an ihre Beobachtungen die Menge der Signalproteine, von denen bekannt ist, dass sie an der Umwandlung von mechanischen in biochemische Signale – der Mechanotransduktion - beteiligt sind. Die zelluläre Antwort kann zu einem veränderten Stoffwechsel oder dem Wachstum des Herzmuskels führen.
„Es zeigte sich, dass alle Knock-out-Mäuse mehr Signalproteine bildeten als die gesunden Tiere“, berichtet Gotthardt. Insbesondere seien jedoch bei den Voll-KO-Mäusen, die mit der Zeit gar kein Titin mehr produzierten, die Proteine FHL2 und p62 verstärkt synthetisiert worden.
FHL2 ist ein Eiweiß, das vor allem an den vorderen, elastischen Bereich des Titins bindet, wo das Molekül wie eine Sprungfeder im Sarkomer verankert ist. Das Protein p62 hingegen wirkt in einem Signalkomplex am hinteren, steiferen Bereich des Titins, wo bei einer Kontraktion des Herzmuskels starke Kräfte entstehen. „Von diesen beiden Proteinen geht ein zusätzliches Wachstumssignal aus, was zu den vergrößerten Herzen der Voll-KO-Mäuse führt“, sagt Gotthardt.
Die Studie liefert eine Grundlage für künftige zielgerichtete Therapien
Der MDC-Forscher und sein Team hoffen, dass ihre Studie dabei hilft, spezifischere Therapien gegen Herzerkrankungen zu entwickeln, die auf einen Defekt im Titin zurückgehen. „Zum einen lassen sich die verschiedenen Titinopathien künftig womöglich besser diagnostizieren und voneinander abgrenzen“, sagt Gotthardt. „Zum anderen besteht eines Tages dann hoffentlich die Möglichkeit, diese Erkrankungen auf den jeweiligen Patienten zugeschnitten zu behandeln.“
Eine Gentherapie, bei der beispielsweise der fehlerhafte Abschnitt im Bauplan für das Riesenmolekül herausgeschnitten und gegebenenfalls ersetzt wird, ist dabei nur eine Option. „Die andere Möglichkeit bestünde darin“, hofft Gotthardt, „die von uns untersuchten Signalwege, die zu einer krankhaften Vergrößerung des Herzens führen, mit kleinen Molekülen ganz gezielt zu verändern.“
Weiterführende Informationen

Literatur
Michael H. Radke et al. (2019): „Deleting Full Length Titin Versus the Titin M-Band Region Leads to Differential Mechanosignaling and Cardiac Phenotypes“. Circulation. doi:10.1161/CIRCULATIONAHA.118.037588



