





Telefon: +49(030)9406-3319
Max-Delbrück-Centrum für
Molekulare Medizin (MDC)
Robert-Rössle-Str. 10
13125 Berlin
Building 27, Room 232
Postal address:
Experimental & Clinical Research Center
Lindenberger Weg 80
13125 Berlin
Building 42-52, Room 1402

We characterize different cardiomyopathy cohorts to identify novel disease genes, to understand the mechanisms for heart failure, and to improve prognosis.
Sabine Klaassen is a member of the task force for the “2023 ESC Guidelines for the management of cardiomyopathies” (Arbelo et al., European Heart Journal, 2023, doi: 10.1093/eurheartj/ehad194).
Sabine Klaassen is co-speaker and principal investigator of the DZHK (Deutsches Zentrum für Herz-Kreislauf-Forschung) partner-site Berlin
The understanding of the natural course and the underlying molecular mechanisms of pediatric CMP and heart failure is incomplete. Therefore, we started the clinical prospective study “Risk evaluation in children and adolescents with primary pediatric cardiomyopathy (RIKADA)” from 01/2013 within the DZHK. The major goals of this project were:
i) definition of disease presentation and progression of pediatric CMP,
ii) genetic characterization of pediatric CMP patients, and
iii) identification of novel genetic causes of pediatric CMP.
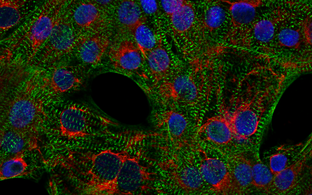
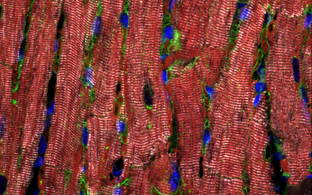
To realize these goals, we established an appropriate workflow involving clinical assessment, in-depth cardiac phenotyping, high-throughput sequencing of patient DNA, and genetic variant analysis. Systematic phenotype and genotype characterization provided important prognostic information in children and adolescents with CMP and their first-degree family members. DCM was the major CMP type, followed by HCM. Most frequently, we detected genetic variants in genes important for sarcomere, desmosome, and z‑disc function, specifically myosin heavy chain 7 (MYH7), myosin binding protein C3 (MYBPC3), cardiac troponin I3 (TNNI3), and desmoplakin (DSP) genes. With targeted high-throughput sequencing at least one pathogenic or likely pathogenic (L)P variant was identified in 38% of the index patients with pediatric CMP (Al-Wakeel-Marquard et al, Journal of the American Heart Association, 2019, doi: 10.1161/JAHA.119.012531; Kühnisch et al., Clinical Genetics, 2019, doi: 10.1111/cge.13645).
This project is part of the DZHK (Deutsches Zentrum für Herz-Kreislauf-Forschung) research at the Pediatric Cardiology Department, DHZC (Deutsches Herzzentrum der Charité).
Pediatric myocarditis and its overlap with dilated cardiomyopathy (DCM) are incompletely understood. Recent analyses from the MYCPEDIG cohort revealed that a substantial subset of children with DCM shows evidence of myocardial inflammation and adverse outcomes. In one-third of these cases, pathogenic variants in CMP-associated genes were identified (Seidel et al., Circulation: Genomic and Precision Medicine, 2021, doi: 10.1161/CIRCGEN.120.003250). Clinically, MYC-DCM patients were significantly younger and had a lower event-free survival compared to the MYC-NonDCM and a primary DCM cohort. These findings suggest that heterozygous DCM causing genetic variants are critical to predict outcome in myocarditis. Our study provides evidence that genetic stratification — particularly in myocarditis cases presenting with a DCM phenotype — is essential for accurate prognosis and tailored management. These phenotype and age-specific findings are useful for personalized management and contributed to revisions of the S2k guideline for myocarditis of the German Society of Pediatric Cardiology. Genetic evaluation in this subgroup of children, newly diagnosed with myocarditis and DCM phenotype should be strongly considered.