
© Klussmann Lab, MDC
Klußmann Lab
Anchored Signalling
Profile
The Anchored Signalling Group aims to elucidate molecular mechanisms conferring specificity to cAMP signalling.
Cyclic adenosine monophosphate (cAMP) is a ubiquitous second messenger with fundamental physiological functions, including the regulation of cardiac contractility, blood pressure and body water homeostasis. To precisely control these distinct mechanisms, cAMP acts in a highly coordinated manner that is specific to the cell type, as well as to time and location.
This compartmentalisation of cAMP signalling involves specific proteins such as A‑kinase anchoring proteins (AKAPs) and phosphodiesterases (PDEs). AKAPs define the composition of cAMP signalling compartments, while PDEs terminate cAMP signalling locally. Dysregulation of compartmentalised cAMP is associated with diseases such as heart failure, hypertension or, if water reabsorption is impaired, diabetes insipidus.
Our goal is to understand the molecular mechanisms that coordinate cAMP signalling in specific cells of the cardiovascular system and to reveal how its dysregulation leads to disease. Based on the components of the signalling cascades identified, we aim to identify novel targets and small-molecule modulators. This could provide new avenues for the development of strategies in treating diseases that involve cAMP signalling compartments where satisfactory treatments do not yet exist. These include heart failure, hypertension, hypertension-induced end-organ injury or water balance disorders such as diabetes insipidus.
Team



Research
A vast range of extracellular cues, such as hormones or neurotransmitters, stimulates the synthesis or release of an intracellular and ubiquitous “second” messenger. Different stimuli can act simultaneously, thereby can even cause an elevation in the same second messenger. This raises the key question of how each distinct stimulus elicits a specific cellular response.
It is now clear that second messengers – such as the cyclic nucleotide, cyclic adenosine monophosphate (cAMP) or Ca2+ – act in manner coordinated in terms of time and location within defined cellular compartments to ensure specificity of cellular responses to each environmental cue. Our group is primarily interested in cAMP signalling compartments.
cAMP predominantly directs cellular processes by integrating inputs from G protein-coupled receptors (GPCRs), which activate the stimulatory G protein (GS). Activated GS induces adenylyl cyclases to generate cAMP. In turn, the cAMP binds to and activates its downstream effector proteins: cAMP-dependent protein kinase A (PKA), exchange proteins activated by cAMP (Epac), cyclic nucleotide-gated ion channels (CNG), hyperpolarisation-activated cyclic nucleotide-gated channels (HCN) and the Popeye domain-containing (POPDC) proteins.
Different GPCRs can, simultaneously, stimulate cAMP synthesis in the same cell. In order to avoid the random activation of cAMP effectors and to enable a specific cellular response to each stimulus, cells spatially restrict the diffusible cAMP and tether its effectors to defined cellular compartments. This compartmentalisation is often achieved by A‑kinase anchoring proteins (AKAPs). At specific cellular sites, AKAPs engage in direct protein-protein interactions with cAMP effectors, their substrates and other signalling proteins, such as kinases and protein phosphatases.
Phosphodiesterases (PDEs) hydrolyse cAMP and are constitutively active. They are also strategically positioned through direct interactions with cellular compartments or protein-protein interactions, including with AKAPs. PDEs thereby locally limit cAMP levels. GS-coupled GPCRs are linked to specifically composed cAMP signalling compartments to facilitate specific cellular responses.
The Anchored Signalling group focuses on compartmentalised cAMP signalling in renal collecting duct principal cells where it controls arginine vasopressin (AVP) ‑mediated water reabsorption, and on cAMP signalling that controls blood pressure in vascular smooth muscle cells (VSMCs) and cardiac myocyte contractility. Through our research, we aim to:
- contribute to a better understanding of the molecular mechanisms underlying compartmentalised cAMP signalling in the cardiovascular system;
- elucidate how the dysregulation of compartmentalised cAMP signalling in specific cells contributes to cardiovascular diseases; and
- identify and validate novel targets for the treatment and prevention of cardiovascular diseases, including in particular diabetes insipidus, hypertension and hypertension-induced cardiac and kidney damage. Innovative therapeutic strategies are urgently required given the ongoing and unmet medical need in these areas.
Projects
Our projects focus on a specific phosphodiesterase (PDE), PDE3A and vasopressin-mediated water reabsorption.
Elucidating functions of PDE3A in the cardiovascular system and targeting PDE3A to protect end-organs from hypertension-induced injury
Hypertension is a major risk factor for secondary cardiovascular complications and diseases, such as cardiac hypertrophy, heart failure, stroke, hypertensive retinopathy or chronic kidney disease (CKD). According to the WHO, an astonishing 1.3 billion people worldwide experience hypertension, leading to 8.5 million deaths from stroke, ischemic heart disease, other vascular and renal diseases each year. 64 million people around the world are affected by heart failure. Only 50% of heart failure patients survive for five years from their diagnosis. These numbers reflect the impact of hypertension and hypertension-induced end-organ damage, not to mention the urgent need for innovative and effective medical therapies to prevent and treat hypertension and related illnesses.
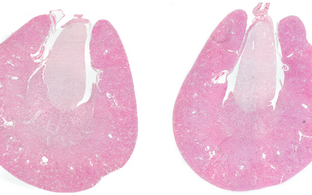
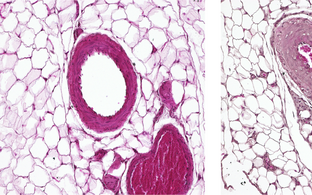
Hypertension with brachydactyly type E (HTNB) is a rare autosomal dominant Mendelian syndrome found in around 20 families to date. HTNB is characterised by harmless brachydactyly type E and progressive severe hypertension that resembles essential hypertension. The severe hypertension is caused by increased proliferation rates of vascular smooth muscle cells (VSMCs) and vessel hyperplasia. HTNB patients’ systolic blood pressure can reach 170 – 250 mmHg and their diastolic pressure 100 – 150 mmHg. Left untreated, patients die of stroke by the age of 50. HTNB causes vascular injury, but surprisingly not the typical end-organ damage induced by hypertension, such as cardiac hypertrophy, heart failure or kidney damage.
We have demonstrated that gain-of-function phosphodiesterase (PDE)3A gene mutations cause HTNB. By examining HTNB patients and studying various cell and animal models, we aim to understand how PDE3A mutations cause hypertension while at the same time protecting organs from hypertension-induced injury. Elucidating these underlying mechanisms could pave the way for novel approaches to the treatment and prevention of hypertension and hypertension-induced end-organ damage.
Links: https://www.ekfs.de/wissenschaftliche-foerderung/aktuelle-foerderungen; https://gepris.dfg.de/gepris/projekt/531907792
Vasopressin-mediated water reabsorption – trafficking water channel aquaporin‑2 (AQP2)
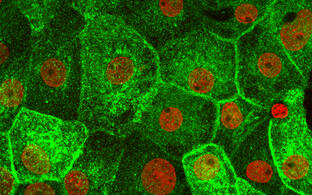
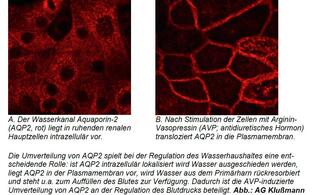
Arginine-vasopressin (AVP; the antidiuretic hormone) induces water reabsorption in renal collecting duct principal cells, thereby fine-tuning body water homeostasis. AVP is secreted by the hypothalamus in response to elevated plasma osmolality, as indicated by thirst. AVP binds to GS-coupled vasopressin type 2 receptors (V2R) on the basolateral plasma membrane of renal collecting duct principal cells, leading to cAMP generation and the activation of protein kinase A (PKA). PKA initiates a signalling cascade that induces the redistribution of water channels, aquaporin (AQP)-2, from intracellular vesicles into the apical plasma membrane.
The insertion of AQP2 introduces water-selective pores into the plasma membrane; water from the lumen of the collecting duct enters through AQP2 along an osmotic gradient and exits the cells through AQP3 and AQP4. The latter two aquaporins constitutively reside in the basolateral plasma membrane. The plasma membrane insertion of AQP2 reduces urine output and decreases plasma osmolality. Dysregulation of the plasma membrane insertion of AQP2 causes diabetes insipidus (DI).
Diabetes insipidus is characterised by massive water loss – up to 20 litres of urine per day – and thirst (polyuria and polydipsia). This water loss can lead to severe complications including seizure, hypotension and hypovolemic shock. Congenital forms of diabetes insipidus, caused by mutations in the genes encoding AVP, V2R or AQP2, affect around 300,000 people worldwide. Meanwhile, drug-induced, acquired forms affect millions of patients: lithium can induce diabetes insipidus in patients with bipolar disorder (1.5 million people in the US alone), while the V2R blocker, tolvaptan, induces diabetes insipidus in patients with autosomal dominant polycystic kidney disease, which affects 12.5 million people worldwide. At this time, there is no satisfactory treatment to reduce water loss in diabetes insipidus.
We have discovered several AKAPs, new kinases and mechanisms that participate in the cAMP-directed control of AVP-mediated water reabsorption. Moreover, we have identified small molecules that promote the plasma membrane localisation of AQP2, which may serve as promising leads for diabetes insipidus treatment. Our group aims to further contribute to the understanding of the molecular mechanisms underlying AVP-mediated water reabsorption and to develop novel small molecules as potential drug candidates for the treatment of diabetes insipidus.
Links: https://www.validierungsfoerderung.de/validierungsprojekte/anti-diainsip; https://gepris.dfg.de/gepris/projekt/530454070
Funding
Our work is kindly supported by individual grants from the Deutsche Forschungsgemeinschaft (DFG), the Federal Ministry of Research, Technology and Space (BMFTR) and the Else Kröner-Fresenius-Stiftung (Key Project), and by the MDC.
Publications
News




Miscellaneous
Dr. Saal van Zwanenbergprijs for Master student Julia Alberts

Martina Schmidt (left) and Julia Alberts (right) at the Dr. Saal van Zwanenberg Prize ceremony
© Hilde de Wolf
On 12 November 2021 the Dr. Saal van Zwanenberg Prize ceremony took place in the Hodson House in Haarlem. Our pharmacy student Julia (M.K.) Alberts was awarded the 3rd Dr Saal van Zwanenberg Prize for excellent Master research in the field of Pharmaceutical Sciences. The Dr. Saal van Zwanenberg Prize is awarded to master students whom master project focused on the identification of drug actions and further improvements for disease treatment. Julia identified in her project that PDE3A inhibitors might act in the future as drug targets to treat cardiovascular disease. Julia aims to enlarge her pharmaceutical network with the price. In the future she aims to work in the pharmaceutical industry with focus on the improvements of regulatory affairs.
Jobs
Open for PhD candidates
For enquires, please contact E. Klussmann and/or
apply through the MDC International PhD program. The program is open for applications twice a year.
Open for MSc and BSc candidates
We are always open for MSc and BSc candidates to carry out the work for their theses in our lab. Projects around PDE3A or the control of AQP2 are available.
For further information please contact enno.klussmann@mdc-berlin.de.
