© Schmitt Lab, MDC
Schmitt Lab
Cancer Genetics and Cellular Stress Responses
Profile
The increasingly faster and detailed molecular dissection of cancer on one side and the rapidly growing list of signaling inhibitors, therapeutic antibodies and so called biologicals (agents that interfere with cellular principles such as proteasomal degradation or histone deacetylation) on the other side are about to make the vision of “personalized cancer precision medicine” a clinical reality, although many obstacles need to be overcome before.
To better understand the underlying mechanism of drug action, signaling networks surrounding the target, and non-cell autonomous implications in the tumor environment, we employ genetically tractable mouse models of cancer and apply integrative “cross-species” bioinformatics as well as focus on conceptually novel – synthetic lethal and “restore & target“ – therapies whose target principles are cell states with their associated vulnerabilities (such as cellular senescence) rather than single molecular lesions.
Team

Research
Mechanistic dissection, functional modeling, and clinical exploitation of DLBCL-derived pathogenetic components (e.g. the BCR/NF-B signaling network) in lymphoma development, CNS tropism, and therapy
(Soyoung Lee, Maurice Reimann, Aitomi Bittner, Animesh Battacharya and collaboration partners (Claus Scheidereit, Jana Wolf, Nikolaus Rajewsky and others)
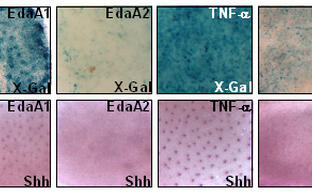
Hyperactivating mutations in the B‑cell receptor (BCR)/NF-B cascade are frequently found in human diffuse large B‑cell lymphoma (DLBCL), and particularly enriched for in those primarily or secondarily localizing to the CNS. NF-B may be oncogenic with respect to cell survival and inflammation, but also tumor-suppressive regarding it’s role in mediating cellular senescence. We modeled distinct DLBCL-derived NF-B mutations in transgenic mouse lymphomas, where they accelerated to various extents Myc-driven lymphoma development. Moreover, we found Eµ-myc transgenic lymphomas with associated CNS manifestation to overexpress an NF-B target gene signature, and, vice versa, distinct NF-B mutants to promote lymphoma CNS tropism. Next, we aim to functionally dissect the NF-B‑governed control of tumor growth, metabolism, cellular stress responses, and non-cell-autonomous implications, and to identify novel vulnerabilities (as therapeutic “Achilles’ Heels”) in reverse genetics models, but also in our “PanOmics”-dissected forward omics-based drug (in)sensitivity model and currently expanded repository of DLBCL material-based patient-derived xenograft (PDX) mouse models (Fig. A). Importantly, “co-clinical trial-like“ functional and multi-omics-based analyses in mouse models accompany a new investigator-initiated trial headed by this group leader and clinical hemato-oncologist, in which previously untreated DLBCL patients receive the Bruton’s tyrosine kinase inhibitor Ibrutinib (Imbruvic®) and the proteasome blocker Bortezomib (Velcade®) as a presumably BCR/NF-B targeting “novel/novel” small compound expansion of the anti-CD20 Rituximab plus CHOP standard immunochemotherapy backbone (termed the “ImbruVeRCHOP” study), and undergo an intense multi-omics-based scientific exploration prior to and during exposure to the trial medication.
The repressive H3K9me3 mark as essential senescence relay and therapeutic target in lymphoma and melanoma
(Yong Yu, Bin Yue, Maja Milanovic Jan R. Dörr, Soyoung Lee, and collaboration partners)
Oncogene-induced senescence (OIS) is a terminal cell-cycle arrest that precludes pre-malignant cells from further expansion via transcriptional silencing of proliferation-related genes in senescence-associated heterochromatin foci (SAHF), which are characterized by the repressive trimethylated histone H3 lysine 9 (H3K9me3) modification. We now found H3K9-active demethylases – namely the Jumonji domain containing 2C (JMJD2C) and the lysine-specific demethylase‑1 (LSD1) – to disable senescence and permit direct transformation under oncogenic Ras or Braf. Accordingly, primary melanoma samples and established melanoma cell lines depend on high H3K9 demethylase activity, and respond to targeted inhibitor therapy in vitro and in vivo with senescence restoration. However, detrimental features of senescent cells, such as associated reprogramming into cancer stemness, asks for their secondary co-therapeutic elimination. Similarly, high-level H3K9-active demethylase expression countered TIS in lymphoma therapy in vitro and in vivo, thereby impairing long-term outcome to chemotherapy. Our data underscore the essential, albeit dynamic role of the H3K9me3 mark in senescence, and unveil the oncogenic potential of H3K9 demethylases as drivers of senescence reversal, thereby providing a mechanistic basis for JMJD2C- or LSD1-targeting strategies in cancer therapy.
Pharmacological restoration and therapeutic targeting of the B‑cell phenotype in classical Hodgkin’s lymphoma
(Yong Yu, Kolja Schleich, Soyoung Lee, and collaboration partners (Jens Peter von Kries, Stephan Mathas)
Classical Hodgkin’s lymphoma (cHL), although originating from B‑cells, is characterized by the virtual lack of gene products whose expression constitutes the B‑cell phenotype. Epigenetic repression of B‑cell-specific genes was previously reported to contribute to the lost B‑cell phenotype in cHL. Restoring the B‑cell phenotype may not only correct a central malignant property, but render cHL susceptible to clinically established antibody therapies targeting B‑cell surface receptors or small compounds interfering with B‑cell receptor (BCR) signaling. In collaboration with the FMP, we conducted a high-throughput pharmacological screening based on more than 28,000 compounds to identify drugs that promote re-expression of the B‑cell phenotype. Intriguingly, restoration of the B‑cell phenotype rendered cHL cells susceptible to clinically approved B‑cell Non-Hodgkin’s lymphoma (B‑NHL)-tailored antibodies (such as anti-CD20 Rituximab) and small compound inhibitors (such as Ibrutinib and Idelalisib) – a conceptually novel “restore & target” strategy.