
Wie pathogene Genvarianten Herzversagen verursachen
Welche molekularen und zellulären Mechanismen bei Menschen mit Kardiomyopathien zu Herzversagen führen, bestimmt die spezifische Genvariante, die der jeweilige Patient oder die jeweilige Patientin in sich trägt. Das ergaben die ersten umfassenden Einzelzell-Analysen von Zellen aus gesunden und kranken Herzen, berichten 53 Forschende aus sechs Ländern in Nordamerika, Europa und Asien in der Fachzeitschrift „Science“.
Je nach genetischer Variante ändern sich die Zusammensetzung der Zelltypen und Profile der Genaktivierung. Mithilfe der Daten könne man gezielte Therapien entwickeln, sagen die Forscher*innen. Diese würden den jeweiligen Gendefekt berücksichtigen, der die Kardiomyopathie des Patienten oder der Patientin verursacht.
Das Team untersuchte 880.000 einzelne Herzzellen

In dieser Illustration wird das kranke Herz als Piñata dargestellt. Es wird von einem DNA-Schläger getroffen, so will die Künstlerin die genetischen Auswirkungen der untersuchten Varianten verdeutlichen. Die Zellen der Patient*innen fliegen wie Konfetti heraus; die Farben erinnern an die fünf analysierten Genotypen.
Die aktiven Gene in rund 880.000 einzelnen Zellen aus 61 erkrankten Herzen und 18 gesunden Referenzherzen zu untersuchen, war ein komplexes Unterfangen. Möglich war das nur in einem interdisziplinären Team. Die Organe haben das Brigham and Woman’s Hospital in Boston, USA, die kanadische University of Alberta und das Herz- und Diabeteszentrum NRW in Bad Oeynhausen, die Ruhr-Universität Bochum und das Imperial College in London, UK, zur Verfügung gestellt.
Zu den Letztautor*innen, die das Projekt geleitet haben, gehören Christine Seidman, Professorin für Medizin und Genetik an der Harvard Medical School und Kardiologin am Brigham and Woman’s Hospital; Jonathan Seidman, Professor für Genetik an der Harvard Medical School; Norbert Hübner, Professor für Herz-Kreislauf- und metabolische Erkrankungen am Max-Delbrück-Centrum für Molekulare Medizin in der Helmholtz-Gemeinschaft (MDC) und an der Charité – Universitätsmedizin Berlin sowie Dr. Gavin Oudit, University of Alberta; Professor Hendrik Milting, Herz- und Diabeteszentrum NRW in Bad Oeynhausen und Ruhr-Universität Bochum; Dr. Matthias Heinig, Helmholtz Munich; Dr. Michela Noseda vom National Heart and Lung Institute am Imperial College London und Professorin Sarah Teichmann, Wellcome Sanger Institute in Cambridge, UK. Die drei Erstautor*innen sind Dr. Daniel Reichart (Harvard), Eric Lindberg und Dr. Henrike Maatz (beide MDC).
Ein Leiden mit zahlreichen Ursachen
Die Forscher*innen haben sich auf die dilatative Kardiomyopathie (DCM) konzentriert. Das ist die häufigste Form der Herzschwäche, die zu Herztransplantationen führt. Bei dieser Krankheit erweitern sich die Wände der Herzkammern (Dilatation), insbesondere im linken Ventrikel – der Herzkammer, die für das Pumpen besonders wichtig ist. Die Muskulatur des Herzens erschlafft, das Herz kann sich weniger gut zusammenziehen und Blut pumpen. Mitunter versagt es ganz. Das Konsortium hat Gewebe von Patient*innen mit verschiedenen Formen erblicher Kardiomyopathien untersucht; die jeweiligen genetischen Veränderungen kommen bei Proteinen mit unterschiedlichen Funktionen im Herzen vor. Die Analysen deuten darauf hin, dass sie auch unterschiedliche Reaktionen auslösen.
Soweit wir wissen, ist es die erste derartige Analyse von Herzgewebe. Wir hoffen, dass dieser Ansatz auch auf andere genetisch bedingte Herzkrankheiten anwendbar ist.
„Wir haben krankheitsauslösende Genvarianten in Herzgewebe auf Einzelzell-Ebene untersucht. So konnten wir präzise kartieren, wie bestimmte pathogene Varianten zu Funktionsstörungen des Herzens führen“, sagt Norbert Hübner, einer der Hauptautoren der Studie. „Soweit wir wissen, ist es die erste derartige Analyse von Herzgewebe. Wir hoffen, dass dieser Ansatz auch auf andere genetisch bedingte Herzkrankheiten anwendbar ist.“
Die Wissenschaftler*innen haben die verschiedenen Mutationen in jedem Herzen genau charakterisiert und sie sowohl untereinander als auch mit gesunden Herzen und solchen, bei denen man die Ursache für die Dilatation nicht kannte, verglichen. Hierfür haben sie sich jeden Zelltyp des Herzens und auch die zahlreichen Subtypen einzeln vorgenommen und mit Methoden der Einzelzellsequenzierung analysiert. Kein Labor könnte die so entstehenden Datenberge allein bewältigen. Nur dank der engen Zusammenarbeit von Spezialisten verschiedener Disziplinen entstand aus Myriaden Mosaiksteinchen ein kohärentes Bild. Die Studie fügt sich zudem in die Arbeit des internationalen Konsortiums zum „Human Cell Atlas“ (HCA) ein, das jeden Zelltyp im menschlichen Körper erfassen und so eine Grundlage schaffen will, um die menschliche Gesundheit zu verstehen und um die Diagnose, Kontrolle und Behandlung von Krankheiten zu verbessern.

Für die Einzelzellsequenzierung müssen die Wissenschaftler zunächst die Zellkerne isolieren. Wenn diese Kerne über einen Mikrofluidik-Chip laufen, werden sie zusammen mit einem Barcode in kleine wässrige Tröpfchen verpackt.

In den Tröpfchen wird der Barcode dann mit der RNA aus dem Zellkern verbunden – also mit den Transkripten, die beim Ablesen der Gene von der DNA entstehen. Auf diese Weise kann die RNA nach der Sequenzierung wieder den einzelnen Zellkernen zugeordnet werden. Die Bilder zeigen Emulsionströpfchen, wie sie sich nach dem Durchlaufen des Chips bilden, mit den blau gefärbten Zellkernen.

In den Tröpfchen wird der Barcode dann mit der RNA aus dem Zellkern verbunden – also mit den Transkripten, die beim Ablesen der Gene von der DNA entstehen. Auf diese Weise kann die RNA nach der Sequenzierung wieder den einzelnen Zellkernen zugeordnet werden. Die Bilder zeigen Emulsionströpfchen, wie sie sich nach dem Durchlaufen des Chips bilden, mit den blau gefärbten Zellkernen.

Algorithmen des maschinellen Lernens ermöglichen eine 2‑D-Darstellung der Genexpressionsprofile. Dabei werden Zellen mit ähnlichen Profilen als Nachbarn dargestellt. So kann man verschiedene Zelltypen voneinander unterscheiden und – wie hier gezeigt – die Zellen des menschlichen Herzens charakterisieren.

„Erst in dieser Auflösung können wir sehen, dass Kardiomyopathien nicht einheitlich immer dieselben pathologischen Signalwege in Gang setzen“, sagt Christine Seidman, eine der Hauptautorinnen. „Vielmehr lösten verschiedene Mutationen jeweils spezifische und einige gemeinsame Reaktionsmuster aus, die zu Herzversagen führen. Diese Mechanismen, die sich je nach Genotyp unterscheiden, zeigen die Ansatzpunkte für die Entwicklung zielgerichteter Therapien.“

Mithilfe neuer Mikroskopie-Technologien kann man die zelluläre Landschaft des Herzgewebes darstellen. Die RNA wird mit fluoreszierenden Molekülen markiert, sodass man Zelltypen unterscheiden kann. Die Grenzflächen der Zellen und die extrazelluläre Matrix sind grün angefärbt, die Zellkerne blau.
Überaktive Bindegewebszellen
Die Zahl der Fibroblasten bleibt gleich. Allerdings werden die bestehenden Zellen aktiver und produzieren mehr extrazelluläre Matrix, die den Raum zwischen den Bindegewebszellen ausfüllt.
„Wir haben zum Beispiel herausgefunden, dass die bei einer DCM auftretende Fibrose – das krankhaft gesteigerte Wachstum von Bindegewebe – nicht deshalb entsteht, weil sich die Fibroblasten des Herzens zu stark vermehren“, sagt Matthias Heinig, der die Daten analysiert hat. „Die Zahl dieser Zellen bleibt gleich. Allerdings werden die bestehenden Zellen aktiver und produzieren mehr extrazelluläre Matrix, die den Raum zwischen den Bindegewebszellen ausfüllt“, ergänzt Eric Lindberg. Es komme somit lediglich zu einer Verschiebung der Subtypen, bei der die Zahl derjenigen Fibroblasten steige, die sich auf die Produktion der extrazellulären Matrix spezialisiert haben.
In den Herzen von Patient*innen mit einem mutierten RBM20-Gen war das Phänomen besonders stark ausgeprägt. Das spiegelte sich auch in der Krankheitsgeschichte wider.
„In den Herzen von Patient*innen mit einem mutierten RBM20-Gen war das Phänomen besonders stark ausgeprägt“, erklärt Henrike Maatz. Dies spiegelte sich auch in der Krankheitsgeschichte wider. Die Betroffenen mussten im Schnitt deutlich früher als Menschen mit einer anderen erblichen Form der DCM ein Spenderherz erhalten, weil ihr eigenes Organ versagt hatte. Mithilfe der Einzelzellsequenzierung sei man auf eine ganze Reihe solcher genotypspezifischer Unterschiede in den erweiterten Herzen gestoßen.
Spezifische Muster von Veränderungen
In den Herzen von Menschen mit arrhythmogener Kardiomyopathie (ACM), die mit gefährlichen Herzrhythmusstörungen verbunden ist, gehen vor allem in der rechten Herzkammer fortschreitend Herzmuskelzellen verloren und werden von Fett- und Bindegewebszellen ersetzt. Auch bei dieser Erkrankung können mehrere Gene verändert sein. In den Analysen hat sich das Team auf das Gen für das Protein Plakophillin 2, kurz PKP2, beschränkt und zelluläre Signalwege, an denen das Protein beteiligt ist, in der rechten und linken Herzkammer miteinander verglichen. Dadurch kann man jetzt beispielsweise besser verstehen, warum sich bei dieser Form der Kardiomyopathie vermehrt Fettzellen bilden.
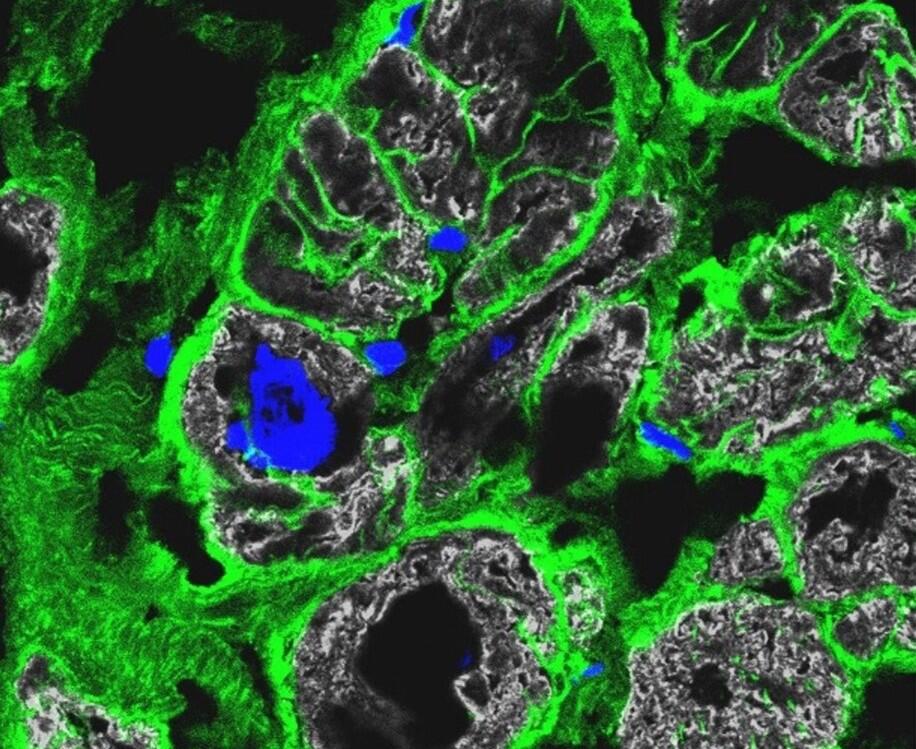
Immunfluoreszenz-Färbung von krankem Herzgewebe: Die Ränder der Zellen sind grün gefärbt, die Zellkerne blau und der Herzmuskel grau.
„Anhand der präzisen molekularen Signaturen, die wir für die hochspezialisierten Zellen des Herzens ermittelt haben, können wir die Kommunikationswege zwischen den Zellen vorhersagen“, sagt Michela Noseda. Je nach genetischer Ursache der Kardiomyopathien komme es zu spezifischen Abweichungen in den zellulären Kommunikationsnetzwerken. „Dies ist ein klarer Beweis dafür, dass ganz spezifische Mechanismen die Krankheit befeuern.“
Aus all diesen Daten haben die Forscherinnen und Forscher schließlich mithilfe künstlicher Intelligenz ein Modell entwickelt. Der Algorithmus kann nun anhand der spezifischen Muster molekularer Veränderungen in den verschiedenen Zelltypen mit großer Wahrscheinlichkeit vorhersagen, um welche Mutation es sich jeweils handelt. Das bestätige, dass pathogene Varianten bestimmter Gene zu Unterschieden in der Gen- und Zellaktivierung führen.
Biomarker für gezielte Therapien

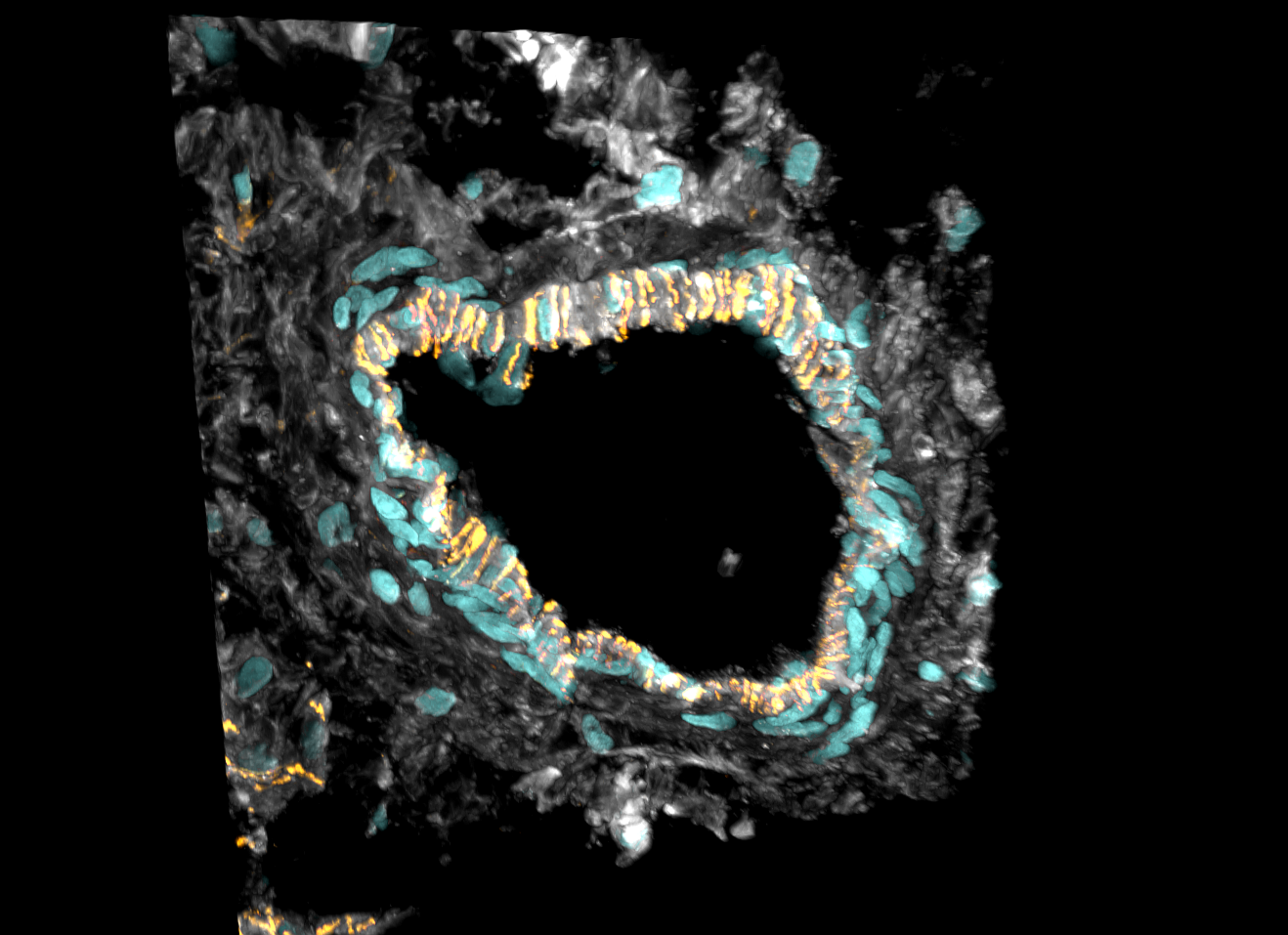
Querschnitt einer Arterie im Herzen: Das Herzgewebe wurde mit Markern für spezifische Zellen angefärbt. Die Endothelzellen, die das Innere der Blutgefäße auskleiden, sind orangefarben, Herzmuskelzellen grau dargestellt. Türkis sind die Zellkerne dargestellt. Dort ist die DNA – die genetische Information – jeder Zelle im Körper gespeichert.
Das langfristige Ziel ist eine personalisierte Therapie von Herzleiden, sagen die Forscher*innen, denn eine genotypspezifische Behandlung wäre effektiver und nebenwirkungsärmer. Um ihrer Vision möglichst schnell näher zu kommen, hat das Konsortium all seine Ergebnisse der Wissenschaft online zugänglich gemacht. Seidman hofft, dass diese Ressource andere Gruppen zu klinischen Studien ermuntert, um neue Behandlungen zur Vorbeugung eines Herzversagens zu entwickeln. Noch sei das eine unheilbare Krankheit.
„Wir haben Gewebe von Patientinnen und Patienten untersucht, die eine Herztransplantation brauchten; es war ihre letzte Option“, sagt Hendrik Milting. „Wir hoffen, dass künftige pharmakologische Therapien das Fortschreiten der Krankheit zumindest verlangsamen können – und dass die Daten aus unserer Studie dazu beitragen.“
Das Herzatlas-Konsortium selbst hat sich derweil seine nächste Aufgabe gestellt. „Das Herzgewebe, das wir untersucht haben, stammte ja von Menschen im Endstadium der Erkrankungen“, sagt Daniel Reichart. „Spannend wird sein, auf welche Veränderungen wir in früheren Stadien stoßen, zum Beispiel auf der Basis von Endomyokard-Biopsien.“ Vielleicht finde man dann auch Biomarker, die eine sehr genaue Diagnose ermöglichen und zugleich den Weg zur besten Therapie weisen, ergänzt Gavin Oudit.
Weiterführende Informationen

- Alle Daten der Studie
- Herzatlas mit Tiefenschärfe (Pressemitteilung)
Literatur
Daniel Reichart, Eric L. Lindberg, Henrike Maatz et al. (2022): „Pathogenic variants damage cell compositions and single cell transcription in cardiomyopathies“. Science, DOI: 10.1126/science.abo1984
Fotos zum Download
- Mithilfe neuer Mikroskopie-Technologien kann man die zelluläre Landschaft des Herzgewebes darstellen. Die RNA wird mit fluoreszierenden Molekülen markiert, sodass man Zelltypen unterscheiden kann. Die Grenzflächen der Zellen und die extrazelluläre Matrix sind grün angefärbt, die Zellkerne blau. © Eric Lindberg, MDC
- In dieser Illustration wird das kranke Herz als Piñata dargestellt. Es wird von einem DNA-Schläger getroffen, so will die Künstlerin die genetischen Auswirkungen der untersuchten Varianten verdeutlichen. Die Zellen der Patient*innen fliegen wie Konfetti heraus; die Farben erinnern an die fünf analysierten Genotypen. © Dr. Eleonora Adami, MDC
- Für die Einzelzellsequenzierung müssen die Wissenschaftler zunächst die Zellkerne isolieren. Wenn diese Kerne über einen Mikrofluidik-Chip laufen, werden sie zusammen mit einem Barcode in kleine wässrige Tröpfchen verpackt. © Eric Lindberg, MDC
- In den Tröpfchen wird der Barcode dann mit der RNA aus dem Zellkern verbunden – also mit den Transkripten, die beim Ablesen der Gene von der DNA entstehen. Auf diese Weise kann die RNA nach der Sequenzierung wieder den einzelnen Zellkernen zugeordnet werden. Die Bilder zeigen Emulsionströpfchen, wie sie sich nach dem Durchlaufen des Chips bilden, mit den blau gefärbten Zellkernen. © Eric Lindberg, MDC | Emulsionströpfchen im Netz | Emulsionströpfchen in Reihe
- Algorithmen des maschinellen Lernens ermöglichen eine 2‑D-Darstellung der Genexpressionsprofile. Dabei werden Zellen mit ähnlichen Profilen als Nachbarn dargestellt. So kann man verschiedene Zelltypen voneinander unterscheiden und – wie hier gezeigt – die Zellen des menschlichen Herzens charakterisieren. © Eric Lindberg, MDC
- Eric Lindberg bei seiner Arbeit im Labor © Felix Petermann, MDC
- Immunfluoreszenz-Färbung von krankem Herzgewebe: Die Ränder der Zellen sind grün gefärbt, die Zellkerne blau und der Herzmuskel grau. © Anissa Viveiros and Dr. Gavin Oudit, University of Alberta
- Querschnitt einer Arterie im Herzen: Das Herzgewebe wurde mit Markern für spezifische Zellen angefärbt. Die Endothelzellen, die das Innere der Blutgefäße auskleiden, sind orangefarben, Herzmuskelzellen grau dargestellt. Türkis sind die Zellkerne dargestellt. Dort ist die DNA – die genetische Information – jeder Zelle im Körper gespeichert. © Sam Barnett and Antonio Manuel Almeida Miranda / Imperial College London
- Ultraschallbild eines kranken Herzens: Beim kranken Herzen ist eine Erweiterung des Ventrikels zu sehen (vergrößertes Kammervolumen), die Kammerwand ist ausgedünnt. Beides sind Anzeichen für eine eingeschränkte Herzfunktion, die bei dilatativer Kardiomyopathie auftritt. © Lech Paluszkiewicz, HDZ-NRW
Kontakte
Prof. Christine Seidman, M.D.
Direktorin des Genetics Center am Brigham and Women’s Hospital
Harvard Medical School
cseidman@genetics.med.harvard.edu
Dennis Nealon
Stellv. Leiter, Media Relations
Harvard Medical School
+1 – 508-494‑6117
Dennis_nealon@hms.harvard.edu
Prof. Norbert Hübner
Leiter der Arbeitsgruppe „Genetik und Genomik von Herz-Kreislauferkrankungen“
Max-Delbrück-Centrum für Molekulare Medizin in der Helmholtz-Gemeinschaft (MDC)
+49 – 30-9406 – 3512 (Sekretariat)
nhuebner@mdc-berlin.de
Jana Schlütter
Redakteurin, Kommunikation
Max-Delbrück-Centrum für Molekulare Medizin in der Helmholtz-Gemeinschaft (MDC)
+49 – 30-9406 – 2121
jana.schluetter@mdc-berlin.de oder presse@mdc-berlin.de
- Max-Delbrück-Centrum für Molekulare Medizin (MDC)
-
Das Max-Delbrück-Centrum für Molekulare Medizin in der Helmholtz-Gemeinschaft gehört zu den international führenden biomedizinischen Forschungszentren. Nobelpreisträger Max Delbrück, geboren in Berlin, war ein Begründer der Molekularbiologie. An den MDC-Standorten in Berlin-Buch und Mitte analysieren Forscher*innen aus rund 60 Ländern das System Mensch – die Grundlagen des Lebens von seinen kleinsten Bausteinen bis zu organübergreifenden Mechanismen. Wenn man versteht, was das dynamische Gleichgewicht in der Zelle, einem Organ oder im ganzen Körper steuert oder stört, kann man Krankheiten vorbeugen, sie früh diagnostizieren und mit passgenauen Therapien stoppen. Die Erkenntnisse der Grundlagenforschung sollen rasch Patient*innen zugutekommen. Das MDC fördert daher Ausgründungen und kooperiert in Netzwerken. Besonders eng sind die Partnerschaften mit der Charité – Universitätsmedizin Berlin im gemeinsamen Experimental and Clinical Research Center (ECRC) und dem Berlin Institute of Health (BIH) in der Charité sowie dem Deutschen Zentrum für Herz-Kreislauf-Forschung (DZHK). Am MDC arbeiten 1600 Menschen. Finanziert wird das 1992 gegründete MDC zu 90 Prozent vom Bund und zu 10 Prozent vom Land Berlin.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}