
Erblicher Bluthochdruck durch überaktives Enzym
Auffällig wurde die türkische Familie aus einem Dorf nahe dem Schwarzen Meer bereits Anfang der Siebzigerjahre. Ein Arzt stellte damals fest, dass bei manchen Mitgliedern der Großfamilie zwei Merkmale stets gemeinsam auftraten: verkürzte Finger und astronomisch erhöhte Brutdruckwerte, zuweilen mehr als doppelt so hoch wie bei gesunden Menschen. Die Betroffenen verstarben in der Regel an einem Schlaganfall, noch bevor sie ihren 50. Geburtstag gefeiert hatten.
Rund zwanzig Jahre später begann eine Gruppe um Professor Friedrich Luft und Dr. Sylvia Bähring am Max-Delbrück-Centrum für Molekulare Medizin in der Helmholtz-Gemeinschaft (MDC), das mysteriöse Phänomen zu erforschen. Es war keine leichte Aufgabe. Erst im Mai 2015 konnten die Forscherinnen und Forscher schließlich im Fachblatt „Nature Genetics“ berichten, dass sie bei allen Patientinnen und Patienten, die an HTNB (Hypertonie mit Brachydaktylie, also Bluthochdruck und Kurzfingrigkeit) litten, auf ein verändertes Gen gestoßen waren. Die Erbkrankheit wird nach seinem türkischen Entdecker auch Bilginturan-Syndrom genannt.
Die Erbanlage kodiert für ein Enzym namens Phosphodiesterase 3A, kurz PDE3A, das sowohl den Blutdruck als indirekt auch das Knochenwachstum reguliert. Die Genmutation, die Luft und sein Team entdeckt hatten, führt dazu, dass das Enzym aktiver ist als gewöhnlich.
Der fehlende Beweis ist nun erbracht

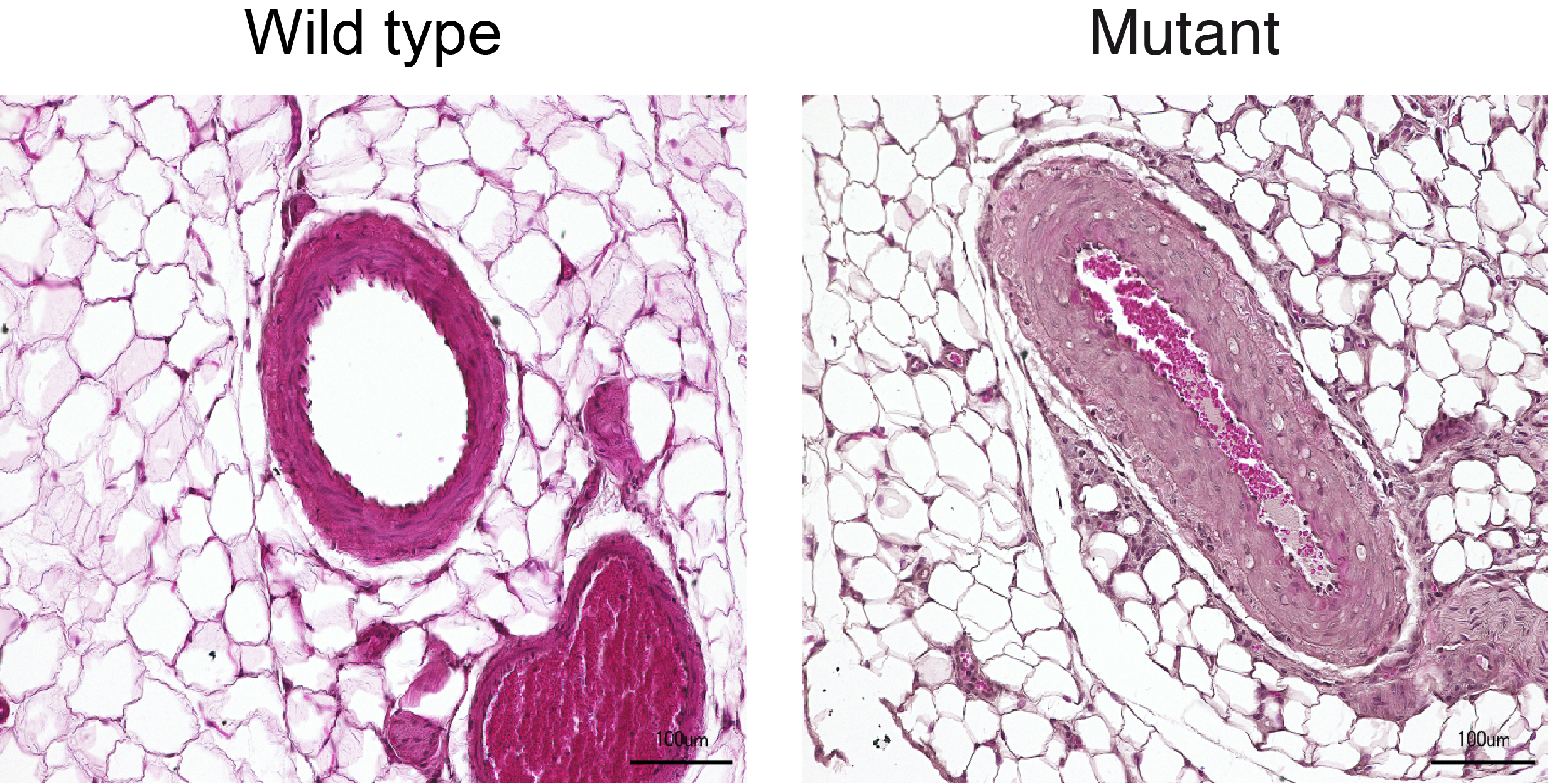
Verengte Mesenterialarterien bei Ratten mit mutiertem PDE3A-Gen (rechts) verursachen durch Erhöhung des Widerstandes den Blutdruck.
Bisher stand allerdings der Beweis aus, dass die mutierte PDE3A wirklich die Ursache für das Bilginturan-Syndrom ist, das man inzwischen auch aus anderen Familien weltweit kennt. Diesen Nachweis hat nun eine internationale Gruppe aus 40 Forscherinnen und Forschern aus Berlin, Bochum, Limburg, Toronto (Kanada) und Auckland (Neuseeland) im Fachblatt „Circulation“ geliefert. An der Studie waren Arbeitsgruppen von MDC und Charité – Universitätsmedizin Berlin beteiligt, darunter Teams um die Professoren Luft, Michael Bader, Maik Gollasch, Dominik Müller, Norbert Hübner, sowie Dr. Arndt Heuser und Dr. Sofia Forslund. Letztautor der Publikation ist Dr. Enno Klußmann, der Leiter der MDC-Arbeitsgruppe „Ankerproteine und Signaltransduktion“.
„Wir haben im Wesentlichen mit zwei Tiermodellen gearbeitet“, berichtet Dr. Lajos Markó, Erstautor neben Maria Ercu. Bei dem einen Modell handelte es sich um genmodifizierte Mäuse, bei denen das menschliche Enzym PDE3A in den Zellen der glatten Muskulatur, aus denen ein Teil der Gefäßwände bestehen, aufgrund der Genveränderung überaktiv war. „Diese Tiere wiesen im Vergleich zu Kontrolltieren einen extrem hohen Blutdruck auf“, sagt der Forscher.
Die genveränderten Ratten litten ebenfalls an der Erbkrankheit
Noch interessanter war für die Wissenschaftler*innen allerdings ein Rattenmodell, das die Arbeitsgruppe von Bader per CRISPR/Cas9-Technik generiert hatte. Das Team hatte mithilfe der Genschere in einer Region des PDE3A-Gens, die bei dem Syndrom mutiert ist, einem sogenannten Mutations-Hotspot, neun Basenpaare verändert. Das daraus hervorgehende Enzym unterschied sich damit in drei Aminosäuren von der gewöhnlichen Variante. „Und wie beim Menschen erhöhte diese winzige Veränderung die Aktivität des Enzyms“, sagt Ercu.

MicroCT Bildgebung der linken Vorderpfoten zeigt, dass im Vergleich zu einer Wildtyp-Ratte (links) die Mittelhandknochen bei Ratten mit mutiertem PDE3A-Gen (rechts) verkürzt sind.
„Die Ratten glichen den menschlichen Patientinnen und Patienten in wirklich sehr erstaunlicher Weise“, ergänzt die Forscherin. „Sie litten nicht nur an hohem Blutdruck, auch die Zehen ihrer Vorderläufe waren deutlich verkürzt – ähnlich wie die Finger bei Menschen mit dem Syndrom.“ Und per Mikro-Computertomographie entdeckten die Forscherinnen und Forscher in den Hirngefäßen der Tiere eine auffällige Schleife, die auch Menschen mit dem Syndrom aufweisen. „Unser Rattenmodell liefert meines Erachtens den endgültigen Beweis dafür, dass das Syndrom durch die Mutation auf dem PDE3A-Gen verursacht wird“, sagt Klußmann.
Ziel ist es, die Volkskrankheit Bluthochdruck effektiver zu behandeln
Man könne nun sogar bereits einen ersten Vorschlag zur Behandlung dieser erblich bedingten Form des Bluthochdrucks machen, ergänzt der Forscher. „Es gibt eine Substanz namens Riociguat, die eigentlich für die Therapie des Lungenhochdrucks zugelassen ist“, sagt Klußmann. Von ihr wisse man, dass sie ein Enzym aktiviert, das einen Botenstoff herstellt, der eine überaktive PDE3A bremsen kann. „Bei Ratten, denen wir ein Derivat von Riociguat verabreicht hatten, sank der Blutdruck auf Normalniveau“, berichtet Klußmann. Zwar seien auch bereits andere PDE3A-Hemmer auf dem Markt, sagt der Forscher, doch aufgrund ihrer Nebenwirkungen seien diese für eine Langzeittherapie eher ungeeignet.
Näher erforschen möchte Klußmann nun noch Interaktionen, die die mutierte PDE3A mit anderen Eiweißmolekülen eingeht. Eine verstärkte Interaktion mit bestimmten Adapterproteinen führe vermutlich dazu, dass sich die Zellen der Gefäßwände stärker vermehren, wodurch sich die Gefäße verengen und der Blutdruck steigt, erläutert der Forscher.
Ein ganz großes Ziel hat Klußmann nämlich noch vor Augen: „Indem wir die Effekte der Interaktionen von PDE3A mit anderen Proteinen besser kennenlernen und verstehen, wie sie an der Regulation des Blutdrucks beteiligt sind, werden wir hoffentlich auch neue und effektivere Therapiemöglichkeiten für die Volkskrankheit Bluthochdruck finden.“
Text: Anke Brodmerkel
Weiterführende Informationen

Downloads
MicroCT Bildgebung der linken Vorderpfoten zeigt, dass im Vergleich zu einer Wildtyp-Ratte (links) die Mittelhandknochen bei Ratten mit mutiertem PDE3A-Gen (rechts) verkürzt sind. Foto: Dr. H. Napieczynska, Heuser Lab, MDC
Verengte Mesenterialarterien bei Ratten mit mutiertem PDE3A-Gen (rechts) verursachen durch Erhöhung des Widerstandes den Blutdruck. Foto: Dr. Q. Fatimunnisa, Bader Lab, MDC
Literatur
Maria Ercu, Lajos Marko et al. (2020): „Phosphodiesterase 3A und Arterial Hypertension“, Circulation, DOI: 10.1161/CIRCULATIONAHA.119.043061
Kontakte
Enno Klußmann
Leiter der Arbeitsgruppe „Ankerproteine und Signaltransduktion“
Max-Delbrück-Centrum für Molekulare Medizin in der Helmholtz-Gemeinschaft (MDC)
+49 (0)30 9406 2596
Enno.klussmann@mdc-berlin.de
Christina Anders
Redakteurin, Kommunikationsabteilung
Max-Delbrück-Centrum für Molekulare Medizin in der Helmholtz-Gemeinschaft (MDC)
+49 (0)30 9406 2118
christina.anders@mdc-berlin.de oder presse@mdc-berlin.de
- Das Max-Delbrück-Centrum für Molekulare Medizin (MDC)
-
Das Max-Delbrück-Centrum für Molekulare Medizin in der Helmholtz-Gemeinschaft gehört zu den international führenden biomedizinischen Forschungszentren. Nobelpreisträger Max Delbrück, geboren in Berlin, war ein Begründer der Molekularbiologie. An den MDC-Standorten in Berlin-Buch und Mitte analysieren Forscher*innen aus rund 60 Ländern das System Mensch – die Grundlagen des Lebens von seinen kleinsten Bausteinen bis zu organübergreifenden Mechanismen. Wenn man versteht, was das dynamische Gleichgewicht in der Zelle, einem Organ oder im ganzen Körper steuert oder stört, kann man Krankheiten vorbeugen, sie früh diagnostizieren und mit passgenauen Therapien stoppen. Die Erkenntnisse der Grundlagenforschung sollen rasch Patient*innen zugutekommen. Das MDC fördert daher Ausgründungen und kooperiert in Netzwerken. Besonders eng sind die Partnerschaften mit der Charité – Universitätsmedizin Berlin im gemeinsamen Experimental and Clinical Research Center (ECRC) und dem Berlin Institute of Health (BIH) in der Charité sowie dem Deutschen Zentrum für Herz-Kreislauf-Forschung (DZHK). Am MDC arbeiten 1600 Menschen. Finanziert wird das 1992 gegründete MDC zu 90 Prozent vom Bund und zu 10 Prozent vom Land Berlin.




{kind=link}
{kind=link}