


















































Experimental and Clinical Research Center
Lindenberger Weg 80
13125 Berlin

Bei Menschen, die an einer genetischen Muskelkrankheit erkrankt sind, ist das aber leider nicht der Fall. Sie erleben eine fortschreitende Abnahme der Muskelkraft und müssen irgendwann den Rollstuhl benutzen. Es gibt einige hundert verschiedene genetische Muskelkrankheiten. Daher sind genetische Muskelkrankheiten keine Seltenheit.
Noch häufiger ist allerdings der Abbau der Muskulatur im Alter, bei chronischen Erkrankungen wie Herz- oder Niereninsuffizienz, bei Krebs oder intensivmedizinischer Behandlung. Der Mechanismus des Muskelabbaus ist heutzutage noch ungeklärt und es gibt keine Therapien.
Weitere Informationen speziell für Patienten finden Sie auf der Seite der Hochschulambulanz für Muskelkrankheiten.
Prof. Dr. Simone Spuler
simone.spuler@charite.de
simone.spuler@mdc-berlin.de
Tel. +49 30 450 540 501÷−504
Monique Wysterski, M.A.
Seit 2020 im Team der AG Spuler
monique.wysterski@charite.de
monique.wysterski@mdc-berlin.de
Tel. +49 30 450 540 504
Dr. rer. nat. Andreas Marg

Seit 2010 im Team der AG Spuler
Fachgebiet: Muskelstammzellen
Aktuelles Projekt: Satellitenzellen und ihre Heterogenität
andreas.marg@charite.de
Tel. +49 30 450 540 524
Dr. rer. nat. Susanne Behr-Perst

Seit 2021 im Team der AG Spuler
Fachgebiet: Projektmanagement Klinischer Studien, Regulatory Affairs, Audit, Qualitäts- und CAPA-Management
Aktuelles Projekt: Leitung der Klinischen Studie MuST
Lebenslauf
susanne.behr-perst@charite.de
Tel. +49 30 450 540 576
Dr. rer. nat. Helena Escobar Fernandez
Seit 2016 im Team der AG Spuler
Fachgebiet: Genombearbeitung, Zelltransplantation, Muskeldystrophie-Mausmodelle
Aktuelles Projekt: Gen-Editierung von Muskeldystrophie, die Mutationen in Patientenzellen verursacht, und neue humanisierte Mausmodelle
helena.escobar@charite.de
helena.escobar@mdc-berlin.de
Tel. +49 30 450 540 526
Dr. med. Elisabetta Gazzerro
Oberärztin der Hochschulambulanz
Seit 2017 im Team der AG Spuler
Fachgebiet: Neuromuskuläre Erkrankungen, Stoffwechselkrankheiten
Aktuelles Projekt: Immunologischer Einfluss bei Muskeldystrophien
elisabetta.gazzerro@charite.de
Tel. +49 30 450 540 514
Dr. rer. nat. Robin Graf
Seit 2021 im Team der AG Spuler
Fachgebiet: Bioingenieurwesen und Biotechnologie
Aktuelles Projekt: Genetische Sicherheit von gen-editierten Muskelstammzellen
robin.graf@charite.de
robin.graf@mdc-berlin.de
Tel.: +49 30 450 540 518
Janine Kieshauer, M.Sc. Biochemie und Molekularbiologie
Seit 2017 im Team der AG Spuler
Fachgebiet: Muskelzellbiologie, regulatorische Angelegenheiten, Entwicklung von Arzneimitteln für neuartige Therapien (ATMP)
Aktuelles Projekt: Validierung der Verarbeitung und Herstellung von menschlichen Muskelstammzellen als ATMP
janine.kieshauer@charite.de
Tel. +49 30 450 540 566
Anne Krause, M.Sc. Molekularbiologie
Seit 2018 im Team der AG Spuler
Fachgebiet: Molekularbiologie, induzierte pluripotente Stammzellen (iPSCs)
Aktuelles Projekt: Gene-Editing von Muskeldystrophie verursachenden Mutationen
anne.krause@charite.de
anne.krause@mdc-berlin.de
Tel. +49 30 450 540 518
Busem Ignak, M.Sc. Molekulare Lebenswissenschaften, Ph.D. Kandidatin
Seit 2021 im Team der AG Spuler
Fachgebiet: Molekulare und zelluläre Biologie, Proteomik, humane induzierte pluripotente Stammzellen und neuronale Differenzierung
Aktuelles Projekt: Präzises Gene-Editing von LGMD2A verursachenden Mutationen
busem.ignak@charite.de
busem.ignak@mdc-berlin.de
Tel. +49 30 450 540 518
Supriya Sai Krishna, M.Sc. Molekulare und Zelluläre Biologie, Ph.D. Kandidatin
Seit 2022 im Team der AG Spuler
Fachgebiet: Molekulare und zelluläre Medizin
Aktuelles Projekt: Präzises Gene-Editing von LGMD2A verursachenden Mutationen
Lebenslauf
supriya.krishna@charite.de
supriya.krishna@mdc-berlin.de
Tel. +49 30 450 540 519
Stephanie Meyer-Liesener
Leitende Technische Assistentin
Seit 2008 im Team der AG Spuler
Fachgebiet: Zellkulturtechniken, Labor-Management
Aktuelles Projekt: Regulation und Fehlregulation von Muskelwachstum
stephanie.meyer@charite.de
Tel. +49 30 450 540 519
Stefanie Haafke, Biologielaborantin
Seit 2012 im Team der AG Spuler
Fachgebiet: Molekularbiologie, Immunfluoreszenzfärbungen, Zellkultur
Aktuelles Projekt: Präzises Gene-Editing von LGMD2A verursachenden Mutationen
stefanie.haafke@charite.de
Tel. +49 30 450 540 518
Student*innen und Praktikant*innen
Oskar Johannssen
April — Juni 2025
A L U M N I
Biniam Bekele, MD, M.Sc.
Im Team der AG Spuler von 2019 — 2022
Ehem. Arzt der Hochschulambulanz und für die MuST-Studie (Klinische Studie) tätig.
Aktuelle Position: Assistenzarzt in Weiterbildung für Herzchirurgie, Klinik für Herz‑, Thorax- und Gefäßchirurgie, Deutsches Herzzentrum der Charité
Dominique Braumann
Im Team der AG Spuler von 2021 – 2022 (Elternzeitvertretung)
Ehem. Projekt: Validierung der Verarbeitung und Herstellung von menschlichen Muskelstammzellen als ATMP
Dr. rer. nat. Silvia Di Francescantonio
Im Team der AG Spuler von 2017 — 2022
Ehem. Projekt: Stammzellbiologie, Muskelbiologie, Quieszenz, Gen-Editing (CRISPR-Cas), Dysferlin
Aktuelle Position: Postdoc-Stipendiatin im Labor von Edgar Gomes am Instituto de Medicina Molecular João Lobo Antunes, Lissabon, Portugal
Dr. rer. nat. Teresa Gerhalter
Im Team der AG Spuler von 2021 — 2022
Ehem. Projekt: suMus — ein digitales Ökosystem rund um die Quantifizierung der Muskelaktivität
Aktuelle Position: Wissenschaftliche Mitarbeiterin am Radiologischen Institut des Uniklinikums Erlangen, Deutschland
Dr. rer. nat. Henning Langer
Im Team der AG Spuler von 2014 — 2018
Ehem. Projekt: Aminosäurestoffwechsel bei Muskelschwund
Aktuelle Position: Postdoc-Stipendiat im Labor von Marcus Goncalves im Weill Cornell Medicine, Center for Metabolic Health in New York, USA
Dr. rer. nat. Jakub Malcher
MyoGrad Doktorandenstipendium von 2013 — 2018; Postdoc 2018 — 2020
Ehem. Projekt: Exon-Skipping und Genome Editing als therapeutische Strategien für Dysferlinopathie
Aktuelle Position: BAYER Co.Lab Programm — und Betriebsleiter
Dr. rer. nat. Eric Metzler
Im Team der AG Spuler von 2015 — 2022
Ehem. Projekt: Entwicklung eines Zell-Therapie-Konzepts basierend auf der Generierung von humanen induzierten Pluripotenten Stammzellen (hiPSCs) und deren Differenzierung in induzierte myogene Zellen
Aktuelle Position: Leitender Wissenschaftler für Stammzellen bei MyoPax (Ausgründung)
Dr. rer. nat. Stefanie Müthel
Im Team der AG Spuler von 2018 — 2024
Ehem. Projekt: Präzises Gene-Editing von LGMD2A verursachenden Mutationen
Aktuelle Position: Teamleitung Forschung & Entwicklung Geneditierung bei LONZA
Adrienne Rothe, Biologisch-Technische Assistentin
Im Team der AG Spuler von 2013 — 2022
Dr. med. Verena Schöwel-Wolf, MBA
Im Team der AG Spuler von 2010 — 2022
Ärztin der Hochschulambulanz für Muskelkrankheiten
Ehem. Projekt: Entwicklung eines präklinischen Dysferlinopathie Modells und Untersuchung des Pathomechanismus und möglicher therapeutischer Strategien; Präklinik und Aufbau der klinischen first-in-human Studie: MuST
Aktuelle Position: CEO von MyoPax (Ausgründung)
Dr. rer. nat. Mark Smith
Im Team der AG Spuler von 2021 – 2022
Christian Stadelmann, M.Sc. Translationale Medizin, Ph.D.
Im Team der AG Spuler von 2020 — 2024
Ehem. Projekt: GMP-konforme Gen-Editierung in primären Muskelstammzellen für die autologe Transplantation
Aktuell: Student der Humanmedizin an der Charité Universitätsmedizin Berlin
Dr. rer. nat. Haicui Wang
Im Team der AG Spuler von 2020 — 2022
Ehem. Projekt: Gen-Editierung in LMNA-verwandten Muskeldystrophie-Patientenzellen
Aktuelle Position: Gruppenleiterin in der Abteilung für Menschliche und Tierische Zellkulturen, Leibniz-Institut DSMZ, Braunschweig, Deutschland
Ehem. Praktikant*innen und Student*innen
Magdalena Bolsinger
Gracia Carrero Peralta
Lucia Link Dopazo
Oskar Johannssen
Leon Kersting
Tim Kühnlenz
Marula Mathew
Luise Minkewitz
Yassin Rassafi
Teresa Schätzl
Leon Zitzelsberger
Helena Escobar Fernandez, Anne Krause, Stefanie Müthel, Christian Stadelmann, Haicui Wang
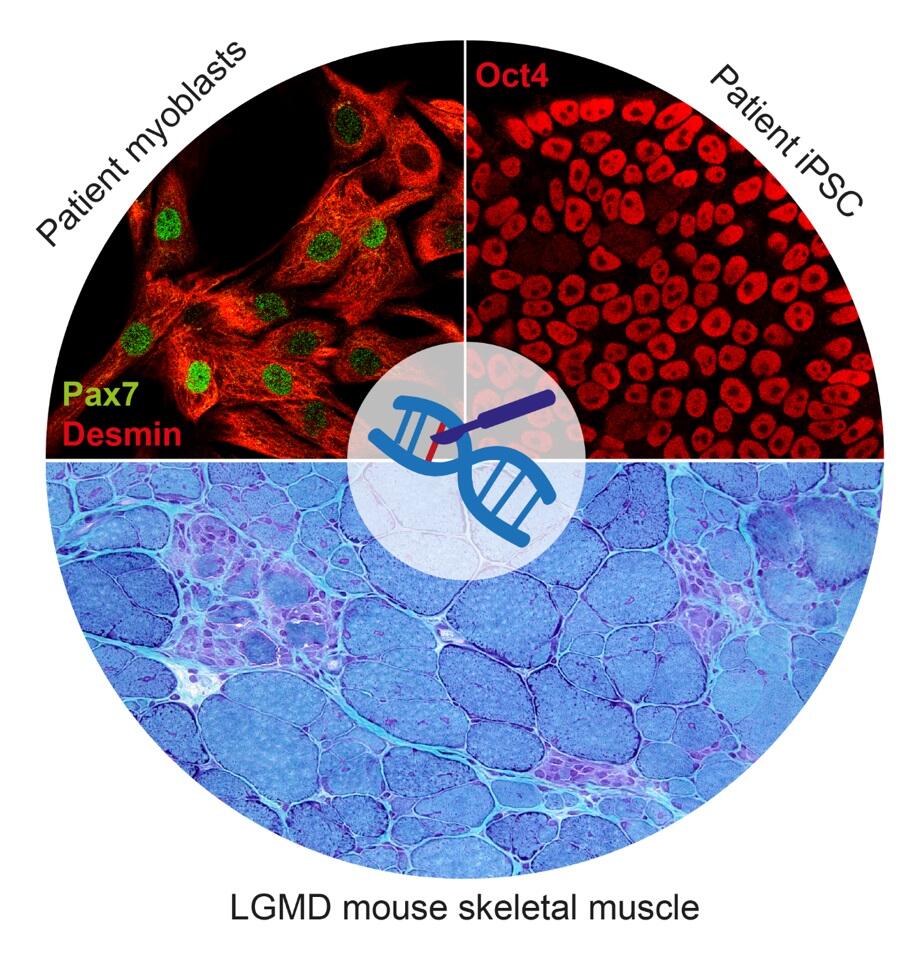
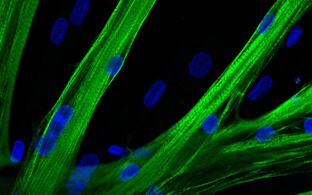
Muskeldystrophien sind progrediente zu Lähmung führende Erkrankungen, für die derzeit keine spezifische Behandlung existiert. Sie sind gekennzeichnet durch fortschreitenden Abbau und eine Degeneration der Skelettmuskulatur, die zu einer Muskelschwäche führen. Diese Erkrankungen sind oft monogen vererbt, d.h. sie werden von Mutationen in einem einzigen Gen hervorgerufen. Ein möglicher therapeutischer Ansatz ist daher, den genetischen Defekt in Zellen, die dem Patienten entnommen werden, zu korrigieren und diese dann für eine autologe Transplantation zu verwenden. Muskelstammzellen (MSC) sind für die Muskelregeneration verantwortlich und wären bei einer Zelltherapie die Zellen der Wahl, um den dystrophen Muskel wiederherzustellen. Allerdings sind sie selten im Muskelgewebe zu finden, haben ein eingeschränktes proliferatives Potential und sind schwierig genetisch zu manipulieren. Deshalb würde die Anzahl der Muskelstammzellen, die entnommen und nach genetischer Korrektur wieder infundiert werden, wahrscheinlich nicht ausreichen, um eine größere Gruppe von Muskeln zu behandeln. Induzierte pluripotente Stammzellen (iPSC) hingegen können aus adulten Körperzellen vom Patienten gewonnen, in Kultur vermehrt, genetisch korrigiert sowie weiter zu Zellen differenziert werden, die einige Eigenschaften von MSC aufweisen. Unsere Arbeit ist fokussiert auf die Entwicklung einer Gene-Editing-Plattform für die Korrektur von Muskeldystrophie-Mutationen in Patienten-eigenen iPSC. Des Weiteren entwickeln wir Methoden, um den Effekt der genetischen Korrektur zuverlässig in vitro und in Transplantationsexperimenten zu testen.
Das direkte Editieren von Genen ist eine vielversprechende Methode, um krankheitsauslösenede Mutationen in Patienten-eigenen Primärzellen zu reparieren. In diesem Projekt wollen wir Mutationen im CAPN3 Gen reparieren. Calpain 3, das Protein, das von CAPN3 kodiert wird, bildet eine Cystein Protease, die vornehmlich im Skelettmuskel exprimiert ist. Mutationen in CAPN3 führen zu Limb Girdle Muscular Dystophy Typ 2 A (LGMD2A), eine progressive Muskelerkrankung, die die weltweit am häufigsten vorkommende LGMD ist und für die es bis heute keine Behandlung gibt.
In unserer Hochschulambulanz behandeln wir mehrere Patienten, die an LGMD2A leiden, und verschiedene Mutationen in CAPN3 aufweisen. Dieses Projekt dient dazu, eine effiziente Plattform für die präzise Geneditierung von krankheitsauslösenden Mutationen in primären humanen Muskelstammzellen aufzubauen. Um die erfolgreiche Genreparatur nachzuweisen, entwickeln wir ebenfalls Methoden um die Wiederherstellung der Genfunktion in vitro und in vivo nachzuweisen. Durch das regenerative Potential von primären humanen Muskelstammzellen, sind wir überzeugt vom Nutzen von reparierten Muskelstammzellen für eine autologe Zelltherapie von LGMD2A-Patienten.
Die Skelettmuskulatur besitzt Muskel-spezifische Stammzellen mit großem regenerativem Potenzial. Sie können nicht nur vom Patienten isoliert, sondern auch außerhalb des Körpers in Kultur vermehrt und manipuliert werden.
Diese Eigenschaften machen die Zellen einen vielversprechenden Kandidaten für die Entwicklung einer autologen Zelltherapie zur Behandlung vererbbarer Muskeldystrophien.
Die heterogene Gruppe von Muskelschwund-Erkrankungen wird häufig durch Mutationen in einem einzelnen Gen ausgelöst. Diese können durch präzise molekulare Werkzeuge, wie der traditionellen CRISPR/Cas9 Genschere oder den neu entwickelten Basen Editoren, korrigiert werden.
Ziel des Projektes ist es Strategien zur Gen-Korrektur zu entwickeln, die sich für eine klinische Anwendung eignen. Hierfür wird der ex-vivo Gene Editing Schritt optimiert, standardisiert und validiert, um eine sichere Anwendung in der Klinik zu gewährleisten.
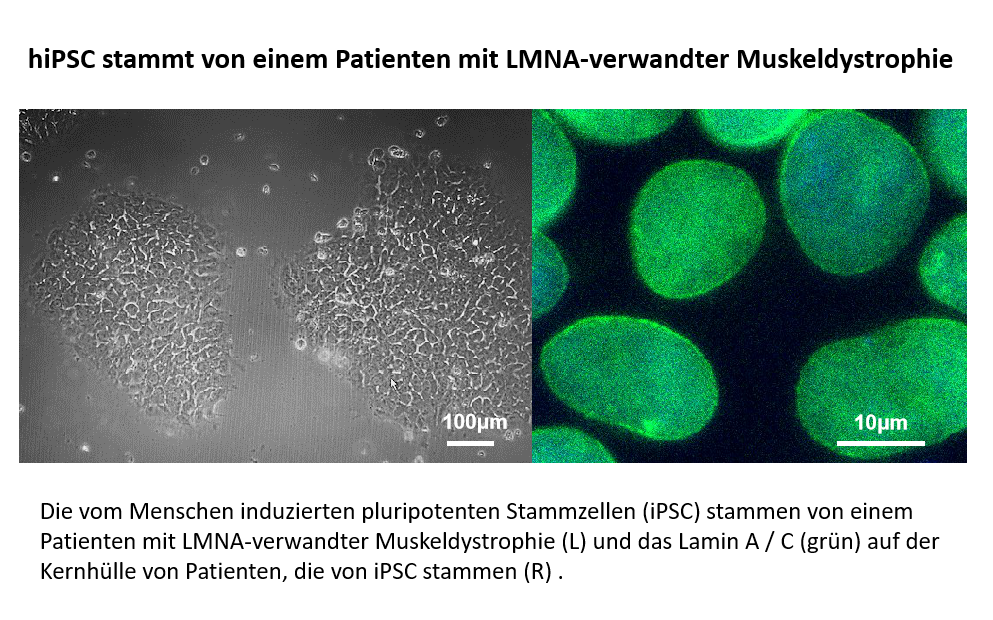
Klassische Laminopathie bezieht sich auf Krankheiten, die durch Mutationen im Gen LMNA verursacht werden, das für Lamin A/C, Schlüsselkomponenten der Kernschicht im Inneren der Kernhülle, kodiert. Der Großteil der klassischen Laminopathie wird durch autosomal dominante LMNA-Mutationen verursacht, und die klinischen Phänotypen können von Muskeldystrophie, erweiterter Kardiomyopathie, Charcot-Marie-Tooth Typ 2B bis hin zu alternder Phänotyp-Progerie variieren.
Wir möchten die neuesten Gen-Editing-Tools verwenden, einschließlich allelspezifischer CRISPR-Cas9- oder CRISPR-Basen-Editing ohne Doppelstrangbrüche, um die Mutationen in Zellen zu korrigieren, die von LMNA-verwandten Muskeldystrophie-Patienten stammen. Das Screening nach dem effizienten Bearbeitungswerkzeug wird mit vom Patienten stammenden induzierten pluripotenten Stammzellen (iPSC) durchgeführt. Die vom Patienten stammenden Muskelstammzellen, die mit der validierten Editierstrategie von iPSC korrigiert wurden, werden weiter für die Transplantationstherapie vorbereitet.
Andreas Marg, Silvia Di Francescantonio, Eric Metzler
Die regenerative Kapazität von Muskelstammzellen (Satellitenzellen) macht diese zu einem optimalen Zelltypen für Geneditierung und zell-basierten Therapien von Muskelerkrankungen. Dafür sollten idealerweise Muskelstammzellen von Patienten isoliert und in vitro manipuliert werden. Weiterhin sollten die Zellen während der Expansion die Charakteristika von Stammzellen nicht verlieren, um für autologe Therapien genutzt werden zu können. Wir möchten eine Zellkulturmethode entwickeln, die es möglich macht, humane primäre Myoblasten in vitro zu manipulieren (z.B. durch Geneditierung) ohne sie aufwendig zu expandieren, da dies zum Verlust der einzigartigen Eigenschaften von Stammzellen führt. Wir können zeigen, dass ein Substrat aus bakterieller Nanocellulose (BNC) Myoblasten für mehrere Wochen bei einer geringen Wachstumsrate hält, ohne dass diese terminal differenzieren. Dadurch ist BNC eine innovative Methode, um langsam teilende Zellen in in vitro Kulturen zu erhalten.
Die CRISPR/Cas9-basierte Geneditierung ist ein leistungsfähiges Werkzeug, um Muskeldystrophie-auslösende Mutationen (z. B. die Gliedergürteldystrophie 2B) zu korrigieren. Trotzdem bleibt die Reparatur von langsam oder nicht-teilenden Zellen eine große Herausforderung. Wir nutzen BNC als Werkzeug um Strategien zur Geneditierung in primären humanen Myoblasten unterteilenden und nicht-teilenden Bedingungen zu vergleichen.
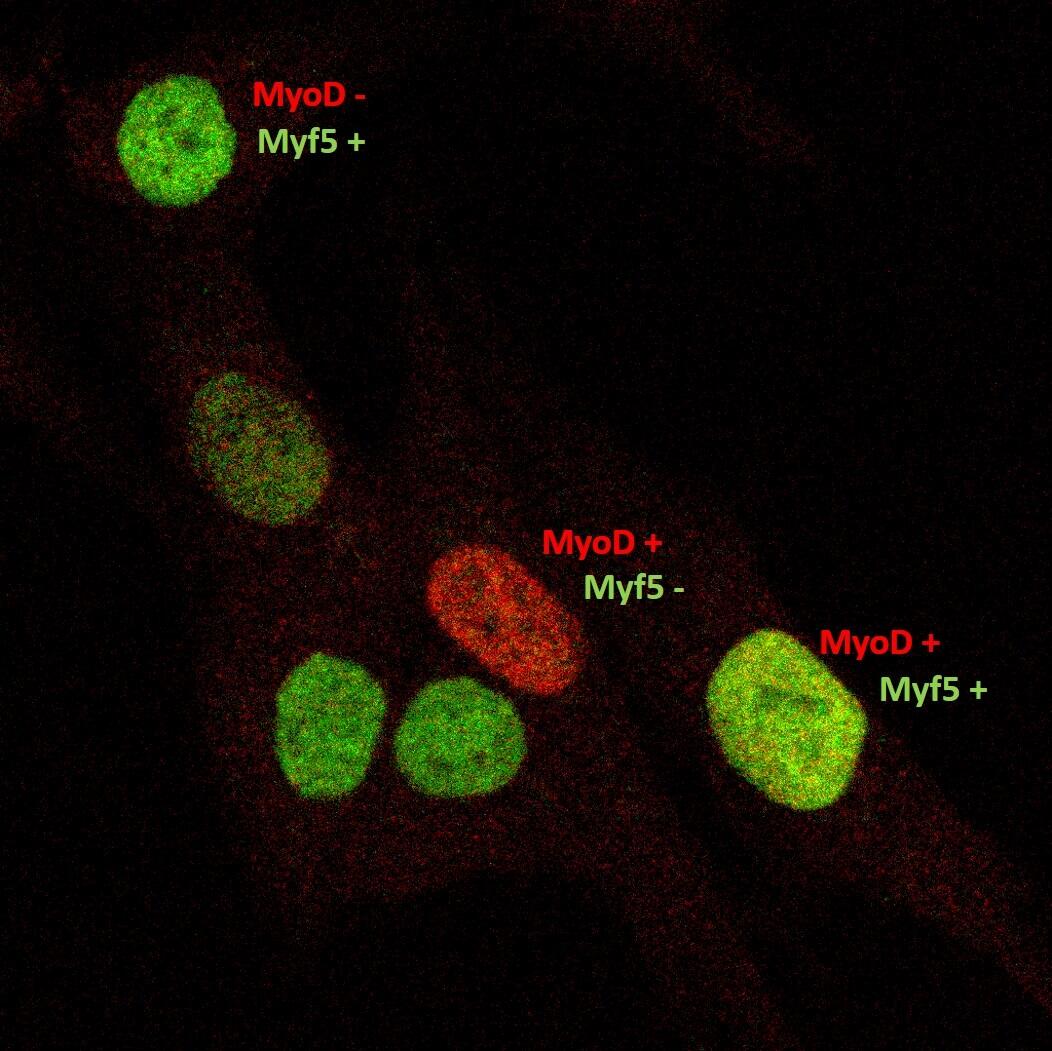
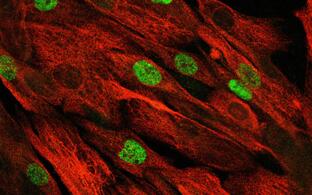
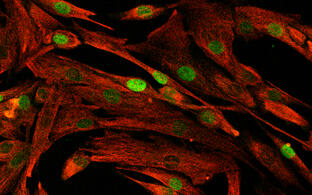
Muskelreparatur und Regeneration erfordern die Aktivierung von Satellitenzellen. Diese seltenen Muskelvorläuferzellen befinden sich in einer speziellen Nische und sind im gesunden Muskel wahrscheinlich mitotisch inaktiv. Es ist auch unklar, in welchem Maße humane Satellitenzellen heterogen sind und sich in ihrer Genexpression, ihrem myogenem Differenzierungspotential und ihren Stammzelleigenschaften unterscheiden. Unser heutiges Verständnis dieser Zellheterogenität ist nur lückenhaft, die klinische Anwendung dieser Stammzellpopulationen erfordert aber ein umfangreiches Wissen auf diesem Gebiet.
In Zusammenarbeit mit dem “Berlin Institute for Medical Systems Biology” (BIMSB) nutzten wir die Drop-seq-Methode für das schnelle Profiling von Tausenden von Einzelzellen. Nach der Sequenzierung erhielten wir das mRNA-Erpressionsprofil von Tausenden von Satellitenzellen.
Auf der Basis dieser Daten versuchen wir nun mit unterschiedlichen Kultivierungs- und Selektionsmethoden die Zellen zu isolieren, die die besten Voraussetzungen für eine erfolgreiche Therapie von Muskeldystrophien haben.
Aktivierte, humane Satellitenzellen sind heterogen
Humane induzierte pluripotente Stammzellen (hiPSCs) stellen den Schlüssel zu einer uneingeschränkten Anzahl autologer Zellen dar, die zur genetischen Korrektur und zur Regeneration von großen Muskeln benötigt werden. hiPSCs wurden bereits aus diversen Zelltypen generiert. Zudem wurden bereits diverse Protokolle zur Generierung von Muskelzellen oder auch Muskelstammzellen aus hiPSCs etabliert. Dennoch ist das biotechnologische und therapeutische Potential dieser aus hiPSCs induzierten Muskelzellen bisher unklar.
Dieses Projekt hat zum Ziel eine effiziente in vitro Differenzierungs-Strategie zu entwickelt inklusive der Frage, ob der somatische Ursprungszelltyp der hiPSCs einen Einfluss auf ihre Kapazität hat in Muskelzellen zu differenzieren. Final sollen induzierte myogene Zellen generiert werden, die ein hohes Potential zur Muskelregeneration in vivo aufweisen, mit dem Ziel eine therapeutische Strategie für Muskeldystrophie-Patienten zu entwickeln.
Joanna Schneider
Critical Illness Myopathie (CIM) ist eine in Folge einer kritischen Erkrankung erworbene Muskelschwäche und eine häufige Komplikation einer intensivmedizinischen Therapie. Sie ist charakterisiert durch eine Muskelatrophie, schlaffe Parese und respiratorische Insuffizienz. Nach Verlassen des Krankenhauses kann diese Muskelschwäche bei einigen Patienten über mehrere Jahre andauern, häufig sogar lebenslang, obwohl alle Risikofaktoren wie Sepsis, Hyperglykämie, Medikamente usw. nicht mehr präsent sind. Wir haben die Hypothese aufgestellt, dass die akute Phase der CIM zur epigenetischen Reprogrammierung von Muskelstammzellen führt, was in einer gestörten Muskelregeneration, einer lang persistierenden Myopathie und einer erhöhten Anzahl doppelsträngiger DNA-Brüche (dsDNA) resultiert. Ziel des Projektes ist es, die innerhalb der ersten Tage nach der Ankunft auf der Intensivstation entstandenen, epigenetischen Veränderungen in Muskelstammzellen von CIM-Patienten zu identifizieren und näher zu charakterisieren. Im Rahmen dieses Projektes führen wir eine Analyse des Epigenoms und Transkriptoms sowie eine Analyse des dsDNA-Bruch-Prozesses der aktivierten Satellitenzellen und frühen Myoblasten von CIM-Patienten durch. Das Projekt ist Teil des Clinical Scientist Programms des Berlin Institut für Gesundheitsforschung und der Charité — Universitätsmedizin Berlin.
Biniam Bekele, Janine Kieshauer, Verena Schöwel-Wolf
Die isolierte Epispadie ist die mildeste Form der Exstrophy-Epispadie-Komplex (EEC); eine angeborene Fehlbildungsstörung, die die Mittellinie der Bauch- und Urogenitalstrukturen betrifft. Patienten mit Epispadie haben einen Defekt im Harnschließmuskel, wo Muskelgewebe durch Bindegewebe ersetzt wird. Daher leiden diese Patienten unter lebenslanger Harninkontinenz mit einer Reihe von medizinischen, psychologischen, sozialen und finanziellen Belastungen. Derzeit gibt es keine kausale Therapie, die diesen Defekt beheben könnte.
Der Skelettmuskel besitzt seine eigenen Stammzellen, die Satellitenzellen. Es handelt sich dabei um hoch regenerative Zellen, die in der Lage sind, sich in Myotuben zu differenzieren und mit vorhandenen Muskelfasern zu fusionieren, um neues Muskelgewebe zu bilden. Unser Labor verfügt über mehrjährige Erfahrung in der Herstellung reiner und hoch regenerativer Satellitenzellpopulationen (PHSats, Primäre humane Satellitenzellen).
Derzeit bereiten wir diese erste Humanstudie vor, bei der PHSats zur Reparatur des Sphinkterdefekts bei isolierten Epispadie-Patienten eingesetzt werden sollen. Wir wollen die Sicherheit und Wirksamkeit unseres Produkts demonstrieren, wobei das Erreichen der Harnkontinenz unser Hauptziel ist. Die Aufnahme des ersten Patienten ist für den Winter 2021⁄22 geplant.
Es handelt sich um eine multizentrische Studie, die in zwei der größten Epispadie-Behandlungszentren Europas durchgeführt wird: der Klinik für Kinderurologie der Universität Ulm und der Klinik für Kinderurologie des Universitätsklinikums Regensburg. Wir stehen auch in engem Kontakt mit der Patientenselbsthilfegruppe Ekstrophie und dem klinischen und wissenschaftlichen Zentrum CURE-Net. Diese Studie wird vom Bundesministerium für Bildung und Forschung finanziert und von der Else-Kröner-Fresenius-Stiftung unterstützt.
Kindgerechte Information zur Studie
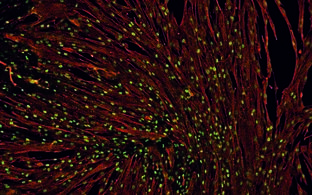
Der Skelettmuskel als größtes Organ des menschlichen Körpers besitzt seine eigene Stammzellpopulation, die sogenannten Satellitenzellen. Die hohe Regenerationsfähigkeit macht die Satellitenzellen zu einer perfekten Quelle für zellbasierte Therapien von Muskelerkrankungen. Wir haben eine neue Technologie entwickelt, die erstmals die millionenfache Vervielfältigung menschlicher Satellitenzellen und gleichzeitig die Verzögerung ihrer Differenzierung in vitro ermöglicht. Wir nennen unser Zellprodukt PHSat (primäres Produkt menschlicher Satellitenzellen). Während der Isolation von PhSats aus Muskelgewebe wird eine Hypothermie-Vorbehandlung genutzt, die ko-isolierte kontaminierende Fibroblasten eliminiert. Ohne die Notwendigkeit einer Zellsortierung sind unsere Zellkolonien zu >98% myogen (desmin-positiv). Das Muskelgewebe wird schonend mechanisch präpariert, eine enzymatische Verdauung findet nicht statt. Auf diese Weise erzeugen wir native (nicht aktivierte) und hoch regenerative Satellitenzellpopulationen. Das regenerative Potenzial von PHSats wurde in präklinischen Wirksamkeitsstudien nachgewiesen: 1. Injizierte PHSats bauen Muskelfasern auf, 2. Sie besiedeln die Nische der Satellitenzellen neu und 3. sie regenerieren den Muskel auch nach einer erneuten Verletzung. Ziel dieses Projektes ist die Entwicklung eines pharmazeutischen Herstellungsprozesses und damit die Überführung des Produktes PHSat in eine klinische Studie. Dieser Prozess muss unter einer spezifischen Infrastruktur erfolgen, wobei höchst standardisierte Parameter festgelegt werden müssen, um das Produkt zur behördlichen Freigabe bestmöglich zu charakterisieren. Dieses konnte durch die Förderung von Pregobio ermöglicht werden.
Momentan befindet sich das Produkt in einer präklinischen Studie, wobei es auf mögliche schädliche Wirkung getestet wird. Diese Daten sind essentiell zum Start der klinischen Studie und wurde durch die Förderung von Spark ermöglicht.
Muskelschwund ist bis heute nicht therapierbar. Allein in der EU sind über 6 Millionen Menschen aufgrund unterschiedlichster Grunderkrankungen davon betroffen. Die Sarkopenie oder genetisch bedingten Muskeldystrophien führen zu einer generalisierten Muskelschwäche. Bei einer Harninkontinenz oder einer Zwerchfellschwäche sind nur einzelne Muskeln nicht funktionsfähig. Trotzdem führt auch so ein isolierter Muskeldefekt zu einer dramatischen Beeinträchtigung der Lebensqualität und kann lebensbedrohlich sein.
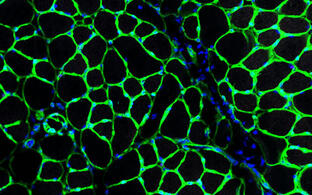
Der Skelettmuskel besitzt seine eigenen Stammzellen, die Satellitenzellen. Sie sind für die hohe Regenerationsfähigkeit des Organs Skelettmuskel verantwortlich. Unsere USP ist die Technologie zur Herstellung hoch regenerativer Satellitenzellen. Unsere Innovation ermöglicht erstmals den hoch standardisierten und effektiven Einsatz dieser Satellitenzellen in der regenerativen Medizin (PHSats, primäre humane Satellitenzellen). Unser Ziel ist die Entwicklung von Satellitenzelltherapien (-/+ Gentechnik) zur Behandlung von Muskelkrankheiten (Abbildung).
Wir haben den ATMP-Markt analysiert und Fallstricke im Rahmen einer ATMP- Produktentwicklung identifiziert. Wir haben eine Strategie zur Stammzelltherapieentwicklung bei Muskelkrankheiten ausgearbeitet. Diese strategische Produktentwicklung wurde im Rahmen des Science4Life Businessplan-Wettbewerbs 2019 ausgezeichnet.
Wir bereiten derzeit eine Erstanwendung eines PHSat Produktes im Menschen vor. Die Vorbereitung der Studie wird durch das Translatorik-Programm der Else-Kröner-Fresenius-Stiftung unterstützt. Ziel ist es hier, einen pränatal unvollständig entwickelten Blasenschließmuskel zu rekonstruieren und eine ansonsten lebenslange Harninkontinenz zu therapieren. Die Finanzierung der Studie ist durch öffentliche Förderung sichergestellt (Bundesministerium für Bildung und Forschung). Der erste Patient soll im Winter 2021⁄22 in die Studie aufgenommen werden.
Darüber hinaus wird die Satelliten-Zelltherapie (primäre und iPS-Zellen) mit Gen-Engineering-Strategien kombiniert und eine Pipeline zur Behandlung einer Vielzahl genetisch bedingter Muskeldystrophien entwickelt.
25.11.2025
Unsere Hochschulambulanz der Charité wird voraussichtlich eines von wenigen Europäischen Studienzentren für eine Therapiestudie bei Myotoner Dystrophie Typ 1 sein.
Zum jetzigen Zeitpunkt können wir Ihnen ein unverbindliches Vorgespräch anbieten. Wenn Sie eine der folgenden drei Fragen mit JA beantworten, wird eine Teilnahme an der Studie nicht möglich sein:
Bitte melden Sie sich bei Interesse unter folgender E‑Mail-Adresse:
DyneStudie-charite@charite.de

Die Coalition to Cure Calpain 3 unterstützt vielversprechende Forschungen zur Entwicklung von Behandlungsmethoden oder Heilmitteln für Gliedergürtel-Muskeldystrophie Typ 2A (LGMD2A/R1, eine Form der Calpainopathie). Die unerbittliche Natur dieser Krankheit führt dazu, dass ihre Betroffenen innerhalb von 11 bis 28 Jahren nach dem Auftreten der Symptome ihre volle Mobilität verlieren und im Rollstuhl sitzen müssen. LGMD2A/R1 zieht deutlich weniger Forschungsgelder an als andere Formen der Muskeldystrophie und daher arbeiten weniger Forscher daran, die Krankheit zu verstehen und ein Heilmittel zu finden.

Die Deutsche Gesellschaft für Muskelkranke e.V. (DGM) ist die größte Selbsthilfeorganisation für Menschen mit neuromuskulären Erkrankungen in Deutschland. Sie wurde 1965 gegründet und bietet umfassende Unterstützung für Betroffene, Angehörige sowie Fachleute. Die DGM setzt sich für die Verbesserung der Lebensqualität von Menschen mit Muskelkrankheiten ein, indem sie Beratung, Information und Vernetzung bietet. Außerdem fördert sie Forschung und Entwicklung neuer Therapieansätze für neuromuskuläre Erkrankungen wie Muskeldystrophien, spinale Muskelatrophien und andere seltene Muskelerkrankungen.

Die Deutsche Muskelschwund-Hilfe e.V. (DMH) ist ein gemeinnütziger Verein, der sich für die Unterstützung von Menschen mit Muskelschwund (neuromuskulären Erkrankungen) einsetzt. Sein Ziel ist es, Betroffenen und ihren Familien zu helfen, indem sie umfassende Beratung, Informationen und Hilfsangebote zur Verfügung stellt. Die DMH fördert zudem Forschungsprojekte zur Entwicklung von Therapien und Heilungsmöglichkeiten für Muskelschwunderkrankungen wie zum Beispiel Muskeldystrophien.Accordion content.

The Jain Foundation is singularly focused on finding a cure for dysferlinopathy, also referred to as LGMD2B, LGMDR2, Miyoshi Myopathy 1

Since 1950, MDA has been the leading philanthropic organization in advancing understanding and treatment of neuromuscular diseases, driving breakthroughs in genetics, diagnostics, and therapies. Our legacy includes ensuring that people living with neuromuscular disease have access to exceptional care from day one of their diagnosis onward, advocating for inclusion, autonomy, and supporting our community through comprehensive educational resources. Through vibrant community programs, we foster deep connections and empower people living with neuromuscular disease to experience the world in the way that they want to, turning challenges into triumphs. Read more about MDA’s journey and the progress we’ve helped make possible. https://www.mda.org/

Strong for Cured Muscles e.V. ist ein gemeinnütziger Verein mit Sitz in Westerstetten, Deutschland. Der Verein setzt sich dafür ein, auf seltene Muskelerkrankungen aufmerksam zu machen und Forschung in diesem Bereich zu unterstützen. Ziel ist es, Therapieansätze zu fördern, die langfristig Heilungsmöglichkeiten für verschiedene Muskelerkrankungen bieten. Darüber hinaus engagiert sich die Organisation auch in der Öffentlichkeitsarbeit und in der Unterstützung von Betroffenen und deren Familien durch Spendenaktionen und andere Initiativen, die das Bewusstsein für neuromuskuläre Erkrankungen schärfen sollen.